
Abstract
Designing useful functionalities in clinically validated, old antibiotics holds promise to provide the most economical solution for the global lack of effective antibiotics, as undoubtedly a serious health threat. Here we show that using the surface chemistry of the cyclodextrin (βCD) cycle and arginine (arg) as a linker, provides more stable ternary antibiotic complex (βCD-arg-cpx). In contrast to classical less stable inclusion complexes, which only modify antibiotic solubility, here-presented ternary complex is more stable and controls drug release. The components of the complex intensify interactions with bacterial membranes and increase the drug’s availability inside bacterial cells, thereby improving its antimicrobial efficacy and safety profile. Multifunctional antibiotics, formulated as drug delivery systems per se, that take the drug to the site of action, maximize its efficacy, and provide optical detectability are envisaged as the future in fighting against infections. Their role as a tool against multiresistant strains remains as interesting challenge open for further research.
Similar content being viewed by others

Introduction
The dwindling of effective antimicrobials poses a very serious threat to global health in the modern world, as the World Health Organization recently pointed out1. Only a few novel antibiotics have reached the market in the last few decades, and no completely new class of antibiotics has been discovered since 19802. With enormous costs and unpredictable and short-term benefits, the discovery of new antibiotics is not a major priority for the pharmaceutical industry2,3. Repurposing, reprofiling, or reusing clinically approved medicines has important advantages in time and cost over discovering new drug candidates, especially in emerging situations such as pandemics4,5. The critical benefits are a predictable safety profile, previous knowledge on manufacturing procedures, established testing protocols, more straightforward regulatory requirements, and shorter bench-to-market periods, among many others6,7. Therefore, it is not surprising that approximately one-third of all approved medicines in the last decade have been repurposed old drugs, which account for 25% of the revenue in the pharmaceutical industry7,8. Much of the effort in the current preclinical antibiotic pipeline is focused on modifying old antibiotics to increase their efficacy, particularly in synergy with other drugs or auxiliary nondrug components9,10. The main scientific challenges in achieving this are the limited penetration, efflux, and toxicity associated with high-dose treatment9,10.
A simple and very effective approach to redesigning old antibiotics includes the formation of instable complexes that modify properties such as solubility, stability, bioavailability, and permeability, thereby directly influencing their therapeutic outcome. In that context, cyclodextrins (CDs) are particularly applicable11,12. With a truncated cone structure, they have a hydrophilic shell (with 7 primary groups oriented toward the narrow edge and 14 secondary sugar hydroxyl groups oriented toward the wider edge of the cone, in the case of β-CD) and a hydrophobic core (with a carbon backbone of 7 glucopyranose units that make up the structure of β-CD) available for interactions with drug molecules11,13,14. Most commonly, antibiotics are formulated as inclusion complexes when the hydrophobic part of the drug interacts with the hydrophobic inner core area of the CD, which consequently increases its solubility by several times11. This approach has been applied to various antibiotics (β-lactams, microlides, fluoroquinolones, sulfonamides, tetracyclines, and aminoglycosides), and their minimal inhibitory concentrations (MICs) were reduced by factors from 2 to more than 10011. It is an enthalpy-driven process, and the host-guest CD-drug complex is in equilibrium with the free drug without chemical bonding12. This approach becomes even more effective if the CD excipient is combined with specially selected auxiliary components (such as hydrophilic polymers, amino acids, or hydroxyl acids) to form ternary complexes15,16,17,18. The hydrophobic part of the drug forms a CD-drug inclusion complex, while the hydrophilic part simultaneously undergoes an acid-base reaction with the auxiliary component to form a salt.
A good example is arginine, a basic amino acid, which forms salts with acidic drugs (e.g., naproxen, zoloprofen, oxaprozin) complexed with CD16,17,18. As a result, an important increase in the stability constant and complexation efficacy is obtained. Therefore, it is expected that combining an antibiotic with CD and arginine into a ternary complex might strongly influence its antimicrobial activity. However, only a few investigations of such complexes have been conducted thus far, among which a study on cefuroxime has shown drastically increased solubility after the formation of ternary complexes with CD and arginine19.
Instead of the less stable inclusion complexes usually formed to increase drug solubility, we designed a more stable antibiotic ternary β-cyclodextrin-arginine-ciprofloxacin (βCD-arg-cpx) complex, in which ciprofloxacin (cpx) is attached to the hydrophilic surface of βCD via an arginine (arg) linker. With this approach, our goal was not simply increasing the solubility of the drug as usual. Here, our synthetized complex system is importantly more stable to control antibiotic release, enable enhanced interactions with the bacterial cell wall and membranes, and provide higher permeability and availability inside the bacteria, consequently improving the efficacy of antibacterial treatment. Along with improved antimicrobial efficacy, considerable improvement in the safety profile was demonstrated.
Results and discussion
CD complex structure and formation
The βCD-arg-cpx complex assembles into well-organized 3D structures (Fig. 1a) as a few micrometers long rods (Fig. 1b) with highly ordered, radial structures composed of laterally connected plates a few tens of nanometers thick (Fig. 1c). A closer look at the single plates and the associated electron diffraction pattern (Fig. 1d) reveals their single-crystalline nature. Morphologically, these assemblies differ from βCD-cpx, and βCD-arg complexes were detected as irregularly shaped, non-assembled particles (Fig. 1e–g).

Illustration of the structure with an arg-cpx crystalline core and βCD at the surface (a). SEM morphology showing long, aligned, rod-like structures of βCD-arg-cpx complex (b) with sharp, nanothick edges (c). Higher-magnification TEM image of the βCD-arg-cpx rods (d) and EDS pattern showing their crystalline nature (insert in (d)). TEM structures revealing differences among βCD-arg-cpx (e), βCD-cpx (f) and βCD-arg (g).
Elemental map** in a single rod βCD-arg-cpx complex (Fig. 2a1) detected C (Fig. 2a2), F (Fig. 2a3), N (Fig. 2a4) and O (Fig. 2a5) elements homogeneously distributed along the large area of a crystal. Further surface composition XPS analysis was performed on cpx drug reference as well as on βCD-arg-cpx complex before and after soft etching with Ar- ions. Since N is present both in antibiotic and arginine (not in CD), while F is present only in cpx, higher N/F ratio in a complex compare to cpx was due to arginine bonding. On the other hand, decreasing C/F and O/F ratios from surface to bulk of the βCD-arg-cpx complex confirmed βCD at the surface. As F- side of cpx is not included in complexation, the maximum at 687.5 eV in F1s spectrum (corresponding to C-F bonds)20 remains unchanged for all three investigated systems (Fig. 2b2). On the other hand, N1s (with two maxima at 401.1 eV and 399.7 eV, corresponding to C-N-C and C-NH bonds20, Fig. 2b4) reveals increased fraction of primarily amines in complex then in cpx free drug which is due to bonding of arginine part. Their increase is observed in bulk area of the complex. The C1s spectrum (with maxima at 287 eV, 285.8 eV and 284.8 eV corresponding to C=O, C-N and C-C, respectively)20 and O1s (with maxima at 533.1 eV and 531.5 eV, corresponding to C-O and (C=O)-OH, respectively)20) show decrease of intensities of maxima belonging to C-O and C-N groups in a complex compare to the free cpx reference which is due to their involvement in complexation.

STEM image (a1) with elemental map** analysis corresponding to carbon (C) (a2), oxygen (O) (a3), nitrogen (N) (a4) and fluorine (F) (a5); XPS analysis of the surface and bulk (obtained after soft etching) of the βCD-arg-cpx complex in comparison to pristine cpx reference- C1s (b1), F1s (b2), O1s (b3) and N1s (b4) high resolution spectra.
The high-aspect-ratio βCD-arg-cpx complex assemblies are highly crystalline, as indicated by their sharp polycrystalline diffraction maxima (Supplementary Fig. 1a). The detected crystal structure is similar to that of the basic deprotonated cpx (as observed in ref. 21), with shifted diffraction maxima and the appearance of new peaks. It also differs slightly from the structure detected for the βCD-NaOH-cpx complex (a reference precipitated by replacing arg with NaOH) and exactly matches the structure of arg-cpx (a reference corresponding to the ciprofloxacin-arginine salt) (Supplementary Fig. 1a), which results from the acid-basic reaction between cpx and arg, formation of the salt and its crystallization. In contrast, crystallization of the separated cpx and arg components within βCD-cpx and βCD-arg complex aggregates is low, giving low intensity and broad diffraction maxima (Supplementary Fig. 1a). Due to the absence of the assembly, the crystal order in these structures is notably decreased. Therefore, the crystalline part of βCD-arg-cpx consisted of arg-cpx salt with an amorphous βCD component associated at the crystal surface.
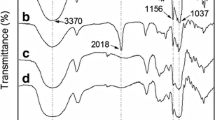
Large structural differences among the different types of complexes were observable particularly in their FTIR spectra (Supplementary Fig. 1b). The FTIR spectrum of the inclusion βCD-arg complex (Supplementary Fig. 1b) showed typical βCD bands with broad O-H stretching at 3300 cm−1 and O-H bending at 1640 cm−1, and C-H stretching at 2925 cm−1, and ring vibration bands as asymmetrical C-O-C and C-C stretching at 1152 cm−1, 1026 cm−1, and 932 cm−1 17,22,23, and guanidine C-N stretching modes from arginine at 1554 cm−1 (more details in Supplementary Fig. 2)24. The intensity of the O-H stretching and bending modes from βCD was altered in the βCD-arg spectrum, but no additional bands were observed (Supplementary Fig. 2). In the βCD-cpx complex, slight position shifts were observed for the bands corresponding to aromatic ring vibrations of cpx and βCD obtained due to the incorporation of the molecule inside the βCD cone, which is typical of inclusion complexes (Supplementary Fig. 3)25,26. In addition, the carboxyl C=O vibrational band of cpx at 1708 cm−1 indicated a neutral molecular form25,26. The βCD-cpx complex did not show any additional bands (Supplementary Fig. 3).
A completely different situation was observed for the βCD-arg-cpx complex (Supplementary Fig. 1b). The stretching carbonyl C=O vibration mode, detected at 1708 cm−1 in βCD-cpx and also in physical mixture of βCD, arg and cpx components, is missing in spectra of both complexes, βCD-arg-cpx and βCD-NaOH-cpx, while complexes’ asymmetric carboxylate vibration (which was missing in case of physical mixture and βCD-cpx) appears at 1578 cm−1 both indicating zwitterion cpx forms within the βCD-arg-cpx and βCD-NaOH-cpx (Supplementary Figs. 3 and S4). In contrast to the spectrum of the inclusion complex βCD-cpx and the spectrum of a physical mixture of components from the complex, in βCD-arg-cpx, the cpx O-H stretching vibration at 3526 cm−1 was missing, another indication of its deprotonated form within the complex. Moreover, typical vibrational bands observed in previous complexes appeared in the spectrum of βCD-arg-cpx with higher intensity, changed intensity ratios, and higher resolution, indicating an increase in the structural order and crystallinity (as observed to carbonyl group vibrations in formulations containing semicrystalline cpx)26. Novel vibrational bands were detected, particularly in the fingerprint area (Supplementary Fig. 4), that did not belong to any of the separate components of the complex (separately and in their physical mixture) but were a consequence of the new complex structure. It was interesting to observe that most of the new bands obtained for βCD-arg-cpx were also detected in arg-cpx and βCD-NaOH-cpx, adjusted to the same pH by using arginine or NaOH, which additionally confirmed the similar structure of complexes formed using these two acidity modulators. However, the presence of arginine within βCD-arg-cpx affected the βCD cycle ring vibrations, observed as the absence of the βCD-typical vibrations at 1079 and 996 cm−1 and additional vibration freedom detected through the new band at 1178 cm−1, which was not detected in βCD-NaOH-cpx. These new interactions within βCD-arg-cpx could be a source of better complex stability, which will be shown later. Investigations of the optical properties of the βCD- complexes enabled further insights into their structural characteristics. The fluorescence emission spectra of the βCD-arg-cpx and arg-cpx complexes (Supplementary Fig. 1c) showed a bathochromic shift when the βCD component was bonded to arg-cpx salt. A shift was observed in both cases when βCD was bonded to the βCD-arg-cpx or βCD-NaOH-cpx complex (Supplementary Fig. 1c). Indirectly, this observation confirmed the presence of this amorphous component on top of deprotonated cpx crystals (detected in XRD and further revealed in FTIR) and provided evidence of the bonding position. The observed fluorescence was assigned to the antibiotic molecule, with piperazinyl electron donor and 4-oxoquinoline-3-carboxylic acid electron acceptor groupsAntimicrobial tests Antibacterial testing was performed using Escherichia coli MG1655 (ATCC 47076), Pseudomonas aeruginosa PAO1 (ATCC 15692), and Staphylococcus aureus Rosenbach (ATCC 12600). The strains were cultured overnight at 37 °C in liquid growth medium (Luria Bertani, LB) (Scharlab, Spain) for E. coli and P. aeruginosa and tryptic soy broth (TSB) (Scharlab, Spain) for S. aureus. Bacterial samples were ultrasonically dispersed in growth medium for 30 s (A = 18%, W = 250 W, on:off = 2:1 s) to form 2 mg/ml stocks, which were further diluted to the tested concentrations. Microtiter plate wells (96-well assay plate, tissue culture-treated polystyrene; Costar 3595, Corning Inc., Corning, NY) were inoculated with 100 μl of bacteria (OD550 = 0.05) and 100 μl of the specific serial dilutions of the tested samples. Controls included growth medium and bacteria without treatment. Incubation was performed in an Infinite M200 Pro multimode microplate reader (Tecan) at 37 °C for 14 h with continuous orbital shaking. Bacterial growth was assessed by measuring optical density at 550 nm every 15 min. All concentrations were tested repeatedly in three replicates (n = 3). To assess bacterial viability, the tested samples were dispersed in LB or TSB growth medium for 30 s (A = 18%, W = 250 W, on:off = 2:1 s) and mixed with bacteria to a final volume of 1 ml (OD550 = 0.3). After incubation for 12 h, 100 μl was centrifuged for 5 min at 6000 rpm, and the supernatant was replaced with 25 μl of Live/Dead BacLight Bacterial Viability Test (Invitrogen, Thermo Fisher Scientific) containing SYTO9 and propidium iodide (PI) in 1X phosphate-buffered saline (PBS) at a 1:1 ratio and a concentration of 3 × 10−6 mg/ml. It was followed by a 15-min incubation in the dark to stain the bacteria. Fluorescent bacteria were visualized by a Nikon inverted fluorescence microscope ECLIPSE Ti-S/L100 (Nikon) coupled with a DS-Qi2 Nikon camera (Nikon). To assess membrane integrity, the tested samples were dispersed in LB or TSB growth medium for 30 s (A = 18%, W = 250 W, on:off = 2:1 s) and mixed with bacteria to a final volume of 1 ml (OD550 = 0.3). After incubation for 12 h, 100 μl was centrifuged for 5 min at 6000 rpm, and the supernatant was replaced with 50 μl of FM 464 (N-(3-triethylammonium propyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide, Invitrogen, Thermo Fisher Scientific)/DAPI (diamidino-2-phenylindole (DAPI; Biotium, Fremont, CA) in Hank’s balanced salt solution (HBSS) (containing 0.4 μl of 5 mg ml-1 FM 464 and 1 μl of 125x DAPI). Samples were stained for 15 min in ice, light protected, and subsequently analyzed using a Nikon fluorescent ECLIPSE Ti-S/L100 microscope. Morphological analyses of bacterial cells affected by the investigated complex were performed using FEISEM (Nova NanoSEM). One hundred microliters of bacteria incubated with the complex for 12 h was centrifuged for 5 min at 6000 rpm and fixed after replacing the growth medium with 50 μl of glutaraldehyde (3 wt %). The fixation step was performed for 3 h at room temperature. Fixed bacteria were deposited on porous membranes by filtration under soft vacuum, washed three times with PBS (for 15 min for each wash), and dehydrated in serially diluted ethanol (30, 50, 70, 90, and 100 wt %, 30 min at each concentration). The samples were dried at critical points. Testing was performed in human lung epithelial A549 cells (ATCC® CCL-185™). Cells were cultured in DMEM/F-12 (Gibco, Thermo Fisher Scientific) supplemented with 1% (v/v) penicillin–streptomycin (Gibco, Thermo Fisher Scientific) and 10% (v/v) decomplemented fetal bovine serum (Gibco, Thermo Fisher Scientific) and grown in a humidified incubator (Memmert) at 37 °C and 5% (v/v) CO2. The tested complex was ultrasonically dispersed in DMEM for 30 s (A = 18%, W = 250 W, on:off = 2:1 s) to form a stock solution (2 mg/ml). Cells grown to confluence in a 96-well plate were treated with serial dilutions of the complex and incubated at 37 °C in 5% CO2 for 24 h. The cytotoxicity assay was performed by adding 20 μl of 10 x Presto blueTM Cell Viability Reagent (Molecular Probes, Invitrogen, Thermo-Fisher Scientific) per well, incubating for 30 min and recording fluorescence at excitation λ = 560 nm and emission λ = 590 nm. The references included the complex without cells in DMEM, the pure dye in DMEM, cells without the complex (as a negative control), and cells with DMSO (as a positive control). All concentrations were tested repeatedly in triplicate (n = 3). Viability has been normalized to cells without treatment (viability % to negative control). Galleria mellonella larvae, used as an in vivo model35, were grown at 34 °C until a 200–250 mg weight. The investigated complex was dispersed in PBS during the toxicity study using ultrasound (30 s, A = 18%, W = 250 W, on:off = 2:1 s) to form a 5 mg/ml stock solution. A 10 μl inoculum of the complex dispersion (in different concentrations) was injected into the upper right proleg area of the larvae using a Hamilton 22-gauge syringe. Each concentration of the complex was injected into five larvae per testing group (n = 5). The control group was inoculated with 10 μl of 1x PBS (Fisher Scientific) in the same manner. After inoculation, the larvae were kept at 37 °C for up to 72 h. Larval mortality was observed every 16–24 h. The testing was done repeatedly. The survival curves were plotted using Kaplan–Meier analysis, and statistically significant differences were determined by the one-sided log-rank test (GraphPad 9.0 Software). During the efficacy study, G. mellonella larvae were preinjected with 10 μl of an infective dose of P. aeruginosa (PAO1) (6.4 × 103 cfu/ml) in the upper right proleg area. An hour after infection, a 10 μl inoculum containing different concentrations of the tested complex dispersed in PBS was injected into the upper left proleg. The following procedure was the same as for the toxicity study. The controls were larvae injected with 1x PBS (negative control) and larvae injected with ciprofloxacin (without complex) at 10 mg/kg (positive control). Each group tested contained five larvae (n = 5). The experiments were done at least in triplicate and repeated 2–3 times (depend on the experiment). Results are presented as mean value ± SD. Differences between groups were assessed by the one-sided log-rank test (GraphPad 9.0 Software). Further information on research design is available in the Nature Research Reporting Summary linked to this article.Live/Dead study in bacteria
FM 464/DAPI study in bacteria
Scanning electron microscopy study in bacteria
In vitro toxicity study
In vivo toxicity
In vivo antibacterial efficacy
Statistics and reproducibility
Reporting summary
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. The source files behind Figs. 2, 4, 5, 6 and 9 are presented in Supplementary Data 1–5 files, respectively. The source files behind Supplementary Figs. 1–4 and 6 are provided in Supplementary Data 6–10 files, respectively.
References
The world is running out of antibiotics, WHO report confirms, World Health Organization (WHO) Report https://www.who.int/news/item/20-09-2017-the-world-is-running-out-of-antibiotics-who-report-confirms (2019).
Plackett, B. No money for new drugs. Nature 586, S50–S53 (2020).
Farha, M. A. & Brown, E. D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 4, 565–577 (2019).
Guy, R. K., DiPaola, R. S., Romanelli, F. & Dutch, R. E. Rapid repurposing of drugs for COVID-19. Science 368, 829–830 (2020).
Zhou, Y., Wang, F., Tang, J., Nussinov, R. & Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2, e667–e676 (2020).
Talevi, A. & Bellera, C. L. Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 15, 397–401 (2020).
Agrawal, P. Advantages and challenges in drug re-profiling. Pharmacovigil S2, e002 (2015).
Naylor, S., Kauppi, D. M. & Schonfeld, J. P. Therapeutic drug repurposing, repositioning and rescue part II: overview. Drug Discov. World 16, 57–72 (2015).
Brown, D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat. Rev. 14, 821–832 (2015).
Theuretzbacher, U., Outterson, K., Engel, A. & Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. 18, 275–285 (2020).
Wong, C. E., Dolzhenko, A. V., Lee, S. M. & Young, D. J. Cyclodextrins: a weapon in the fight against antimicrobial resistance. J. Molec. Eng. Mater. 5, 1740006 (2017).
Saokham, P., Muankaew, C., Jansook, P. & Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 23, 1161 (2018).
Uekama, K., Hirayama, F. & Irie, T. Cyclodextrin drug carrier systems. Chem. Rev. 98, 2045–2076 (1998).
Nardello-Rataj, V. & Leclercq, L. Encapsulation of biocides by cyclodextrins: toward synergistic effects against pathogens. Beilstein J. Org. Chem. 10, 2603–2622 (2014).
Redenti, E., Szente, L. & Szejtli, J. Cyclodextrin complexes of salts of acidic drugs. Thermodynamic properties, structural features, and pharmaceutical applications. J. Pharm. Sci. 90, 979–986 (2001).
Mura, P. et al. Solid-state characterization and dissolution properties of naproxen–arginine–hydroxypropyl-b-cyclodextrin ternary system. Eur. J. Pharm. Biopharm. 59, 99–106 (2005).
Sherje, A. P., Patel, F., Murahari, M., Suvarna, V. & Patel, K. Study on effect of L-arginine on solubility and dissolution of Zaltoprofen: preparation and characterization of binary and ternary cyclodextrin inclusion complexes. Chem. Phys. Lett. 694, 120–128 (2018).
Mennini, N., Maestrelli, F., Cirri, M. & Mura, P. Analysis of physicochemical properties of ternary systems ofoxaprozin with randomly methylated-ß-cyclodextrin and l-arginine aimed to improve the drug solubility. J. Pharm. Biomed. Anal. 129, 350–358 (2016).
Sapte, S. & Pore, Y. Inclusion complexes of cefuroximeaxetil with β-cyclodextrin: physicochemical characterization, molecular modeling and effect of L-arginine on complexation. J. Pharm. Anal. 6, 300–306 (2016).
Rincón-Ortiz, S. A., Botero, M. A. & Ospina, R. XPS characterization of ciprofloxacin tablet. Surf. Sci. Spectra 29, 014020 (2022).
Al-Omar, M. A. Ciprofloxacin: physical profile. Profiles Drug Subst. Excip. 31, 163–178 (2004).
Sambasevam, K. P., Mohamad, S., Sarih, N. M. & Ismail, N. A. Synthesis and Characterization of the Inclusion Complex of β-cyclodextrin and Azomethine. Int. J. Mol. Sci. 14, 3671–3682 (2013).
Kacso, I., Borodi, G., Fărcaş, S. I. & Bratu, I. Inclusion compound of vitamin B13 in β-cyclodextrin. structural investigations. J. Phys.: Conf. Ser. 182, 012009 (2009).
Sosa, L. V. et al. The structure, molecular dynamics, and energetics of Centrin-Melittin complex. Proteins 79, 3132–3143 (2011).
Bhongade, B., Talath, S. & Dhaneshwar, S. A validated method for the quantitation of ciprofloxacin hydrochloride using diffuse reflectance infrared fourier transform spectroscopy. Int. J. Spectrosc. ID 2014, 294612 (2014).
Mesallati, H., Umerska, A., Paluch, K. J. & Tajber, L. Amorphous polymeric drug salts as ionic solid dispersion forms of ciprofloxacin. Mol. Pharmaceutics 14, 2209–2223 (2017).
**e, S. et al. Design and synthesis of theranostic antibiotic nanodrugs that display enhanced antibacterial activity and luminescence. Proc. Natl Acad. Sci. USA 114, 8464–8469 (2017).
Neves, M. A. C., Yeager, M. & Abagyan, R. Unusual arginine formations in protein function and assembly: rings, strings, and stacks. J. Phys. Chem. B 116, 7006–7013 (2012).
Zhang, F., Gu, S., Ding, Y., Zhang, Z. & Li, L. A novel sensor based on electropolymerization of β-cyclodextrin and l-arginine on carbon paste electrode for determination of fluoroquinolones. Anal. Chem. Acta 770, 53–61 (2013).
Hooper, D. C. Mechanisms of fluoroquinolone resistance. Drug Resist. Updat. 2, 38–55 (1999).
Bos, J. et al. Emergence of antibiotic resistance from multinucleated bacterial filaments. PNAS 112, 178–183 (2015).
Turnbull, L. et al. Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Comm. 7, 11220 (2016).
Moya-Andérico, L. et al. Utility of Galleria mellonella larvae for evaluating nanoparticle toxicology. Chemosphere 266, 129235 (2021).
Blanco-Cabra, N. et al. Novel oleanolic and maslinic acid derivatives as a promising treatment against bacterial biofilm in nosocomial infections: an in vitro and in vivo study. ACS Infect. Dis. 5, 1581–1589 (2019).
Moya-Andérico, L., Admella, J., Fernandes, R. & Torrents, E. Monitoring gene expression during a Galleria mellonella bacterial infection. Microorganisms 8, 1798 (2020).
Acknowledgements
The authors are grateful to Angel Blanco Blanes and Dr. Samuel Sanchez from the Smart Nano-Bio-Devices group at the Institute for Bioengineering of Catalonia for zeta potential measurements as well as to Dr Dušan Žigon from Center for Mass Spectrometry at Jozef Stefan Institute for ToF MS analysis. The European Commission has funded this work under Horizon 2020’s Marie Skłodowska-Curie Actions COFUND scheme (Grant Agreement no. 712754) and by the Severo Ochoa program of the Spanish Ministry of Science and Competitiveness (Grant SEV-2014-0425 (2015-2019)). E.T. was supported by grants from the Spanish Ministerio de Economia y Competitividad (MINECO/FEDER) (RTI2018-098573-B-100), Generalitat de Catalunya (2017SGR-1079 and CERCA program), the Catalan Cystic Fibrosis associations and the La Caixa Foundation. Additional funding support has been provided to M.V. by grants from the Slovenian Research Agency (ARRS) (grants J2-8169, N2-0150, and P2-0091).
Author information
Authors and Affiliations
Contributions
M.V. research concept, synthesis and characterization, writing first draft; L.G. XRD and FTIR analyses, M.K. TEM and EDS analyses, J.H. STEM and elemental map** analysis, B.J. XPS analysis, L.M.-A. in vivo study, M.C. in vitro toxicity, E.T. mentoring and funding. All authors contributed to editing text to final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks John-Sigurd Svendsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vukomanovic, M., Gazvoda, L., Kurtjak, M. et al. Development of a ternary cyclodextrin–arginine–ciprofloxacin antimicrobial complex with enhanced stability. Commun Biol 5, 1234 (2022). https://doi.org/10.1038/s42003-022-04197-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-04197-9
- Springer Nature Limited