
Abstract
In the current study, we aimed to investigate whether disulfiram (DSF) exerts a neuroprotective role in cerebral ischemiareperfusion (CI-RI) injury by modulating ferredoxin 1 (FDX1) to regulate copper ion (Cu) levels and inhibiting inflammatory responses. To simulate CI-RI, a transient middle cerebral artery occlusion (tMCAO) model in C57/BL6 mice was employed. Mice were administered with or without DSF before and after tMCAO. Changes in infarct volume after tMCAO were observed using TTC staining. Nissl staining and hematoxylin–eosin (he) staining were used to observe the morphological changes of nerve cells at the microscopic level. The inhibitory effect of DSF on initial inflammation was verified by TUNEL assay, apoptosis-related protein detection and iron concentration detection. FDX1 is the main regulatory protein of copper death, and the occurrence of copper death will lead to the increase of HSP70 stress and inflammatory response. Cuproptosis-related proteins and downstream inflammatory factors were detected by western blotting, immunofluorescence staining, and immunohistochemistry. The content of copper ions was detected using a specific kit, while electron microscopy was employed to examine mitochondrial changes. We found that DSF reduced the cerebral infarction volume, regulated the expression of cuproptosis-related proteins, and modulated copper content through down regulation of FDX1 expression. Moreover, DSF inhibited the HSP70/TLR-4/NLRP3 signaling pathway. Collectively, DSF could regulate Cu homeostasis by inhibiting FDX1, acting on the HSP70/TLR4/NLRP3 pathway to alleviate CI/RI. Accordingly, DSF could mitigate inflammatory responses and safeguard mitochondrial integrity, yielding novel therapeutic targets and mechanisms for the clinical management of ischemia–reperfusion injury.
Similar content being viewed by others
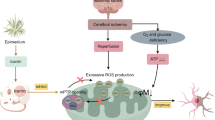


Introduction
The incidence and mortality rates of cerebral ischemic disease are gradually increasing worldwide. Moreover, cerebral ischemic disease is the most common cause of permanent disability in adults1. Thrombolysis/thrombolysis is the most effective treatment for ischemic stroke. Since 2015, substantial advancements have been made in research exploring the treatment of acute ischemic stroke, leading to an expanded time window for emergency endovascular intervention, from the initial 6 h to a more extended period of 24 h2. Following cerebral ischemia, reperfusion of blood flow induces a cascade of injurious effects, commonly referred to as cerebral ischemia/reperfusion injury (CI/RI). This process involves various types of regulated cell death (RCD) and neuroinflammatory responses, considerably contributing to its progression3,4. Mitigating the adverse effects of reperfusion has been a persistent focal point for clinicians striving for breakthroughs.
In living organisms, cells exposed to extreme physicochemical or mechanical stressors may undergo an immediate and uncontrolled structural collapse, a phenomenon known as accidental cell death. Conversely, RCD involves specific signaling cascades and molecular-defined effector mechanisms, encompassing fundamental processes, such as organogenesis and tissue remodeling, eliminating unnecessary structures or cells, and controlling cell numbers. Additionally, RCD can be triggered by exogenous perturbations in the intracellular or extracellular microenvironments5,6. Currently, more than 10 types of RCDs have been identified, encompassing a diverse range of non-apoptotic RCD modalities, such as autophagy, pyroptosis, ferroptosis, and endogenous cell demise, in addition to the conventional apoptotic pathway7,8. In contrast to apoptosis, these RCD pathways elicit inflammatory responses within the body and have been increasingly associated with cancer9
CI/RI induces a robust inflammatory response initiated by damage-associated molecular patterns (DAMPs) released from injured cells, including signaling molecules, such as adenosine, heat shock proteins (HSPs), high-mobility group protein B1 (HMGB1), and interleukin (IL)10,11,12. In a healthy central nervous system, various types of DAMPs are expressed and released following injury to activate inflammatory signaling pathways11. The aforementioned DAMPs use pattern recognition receptors (PRRs) to initiate and augment immune responses13. Extracellular HSP70 is a typical DAMP14 PRRs are predominantly expressed in astrocytes and microglia, enabling the detection of pathogen-derived or endogenous ligand release. Toll-like receptors (TLRs) of the PRR family are an example of this phenomenon15. TLR4 plays a pivotal role and serves as the primary receptor for HMGB116. In addition, HSP70 regulates TLR417,18 TLR4 is most commonly associated with the expression and release of IL-1 and tumor necrosis factor-α (TNF-α), and this association typically occurs in a ligand-dependent manner. Nuclear factor (NF)-κB, positioned downstream of the TLR4 signaling pathway, plays a pivotal role in orchestrating immune responses, cellular proliferation, and differentiation. Upon activation, TLR4 induces nuclear translocation of NF-κB (p65), thereby facilitating the expression of diverse inflammatory cytokines, such as IL-1β and TNF-α19. The activation of the TLR4/NF-κB signaling pathway facilitates the assembly of a complex between NLRP3 and apoptosis-associated speck-like protein with a CARD (ASC), which subsequently interacts with the cysteine protease caspase-1 to form inflammasomes20,21. The classic pyroptotic pathway, mediated by the NLRP3 inflammasome, is pivotal in determining functional outcomes following stroke22.
Recently, a novel RCD pattern, distinct from the well-known RCD pattern, has been identified and designated as “cuproptosis”. This unique mode of cell death relies on the copper (Cu)-mediated targeting of lipoacylated tricarboxylic acid (TCA) cycle proteins and is strongly associated with mitochondrial respiration23. FDX1 converts Cu (II) to the highly toxic Cu (I), resulting in the aggregation of fatty acylated proteins, exhaustion of iron-sulfur cluster proteins, HSP70 activation, and induction of intracellular toxic oxidative stress, ultimately causing cell death. Importantly, this process is associated with mitochondrial respiration. A meta-analysis has revealed that serum Cu levels are substantially elevated during the acute phase of stroke24. Recent evidence suggests that Cu ions participate in various transformation mechanisms that damage brain tissue during fusion injury25. The induced cuproptosis activates HSP70 to initiate an immune inflammatory response.
Disulfiram (DSF) is a well-established anti-alcoholic medication scientifically validated for its safety and potential in targeted tumor therapy26,27. Recent research has demonstrated the potential of DSF in inhibiting multiple inflammatory reactions and regulating inflammation-related targets, highlighting its potential as an anti-inflammatory agent3A).

Mitochondrial and myelin morphological changes and copper ion detection. (A) Representative transmission electron micrographs showing the brain tissues. Mitochondria (blue arrows) were obviously swollen and cristae was broken and disappeared after tMCAO. Mitochondria were relatively intact in DSF+24h-tMCAO group. Dark black is the nerve myelin sheath (white arrows), 24 h-tMCAO group. The phospholipid layer is loosely wrapped and thinned in thickness. Myelin sheath were relatively intact in DSF+24 h-tMCAO group. (B) Copper ion levels decrease with DSF (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; and ns not significant.
Discussion
Herein, our findings suggest that DSF can exert inhibitory effects on the inflammatory response and cell death induced by CI/RI via the modulation of the TLR4/NLRP3/NF-κB pathway and FDX1/TCA cycle, thereby affording a neuroprotective role. Treatment with DSF alone could suppress the expression of FDX1/TCA cycle-related proteins and the HSP70/TLR4/NLRP3 pathway during CI-RI to protect nerves and inhibit inflammation.
Cu plays an indispensable role in the physiological processes of the central nervous system. The disruption of Cu homeostasis can exert a multitude of detrimental effects on brain development and function. Compared with other organs, the brain exhibits the highest Cu concentration, second only to the liver. Notably, the average Cu content in the globus pallidus surpasses that in the liver35. As a cofactor, Cu plays an indispensable role as a reducing agent in the catalytic activity of peptidyl-glycine alpha-amide monooxygenase, dopamine β-monooxygenase, Cu-zinc (Zn) superoxide dismutase, and ceruloplasmin36. The presence of Cu is crucial for catecholamine biosynthesis, neuropeptide activation, and the regulation of mitochondrial function37. However, excess Cu induces lipid deposition through oxidative stress, mitochondrial dysfunction38 promotes neurodegenerative changes, and cell death39 Cu transporters SLC31A1 (CTR1), ATP7A, and ATP7B are known to regulate the Cu content in cellular compartments and maintain Cu homeostasis. CTR1, a master regulator of Cu uptake, acts on the plasma membrane, and the genetic inactivation of CTR1 results in Cu deficiency in cells40. In the choroid plexus (ChPl), Cu flows from the epithelial cells of the ChPl into the cerebrospinal fluid (CSF). In patients with Menkes disease, ATP7A inactivation is known to cause Cu deficiency in the brain, numerous metabolic abnormalities (including catecholamine imbalance), delayed neurodevelopment, and death during early childhood41. Generally, ATP7B transports Cu from the cytosol into the lumen of the secretory pathway for incorporation into Cu-dependent enzymes and traps excess Cu in vesicles for further export from the cell. The precise impact of these activities on cellular metabolism depends on the specific cell type and can exhibit significant variation across different tissues42. Therefore, the negative effects of ATP7B inactivation on brain metabolism are evident in Wilson’s disease. Copper-induced protein transport has been described as a key feature of copper-ATPase, and the central link of this mechanism is the transfer of ATP7A and ATP7B from TGN to peripheral or cytoplasmic vesicles, respectively. The displacement of these proteins is specifically triggered by elevated levels of their own ligand, Cu (I), not Cu (II), and is intended to efflux to restore intracellular copper levels43,44,45 FDX1 helps convert Cu (II) to the more toxic Cu (I), and in our study, DSF inhibited FDX1 expression, reduced Cu (I) levels, and reduced transport of ATP7A and ATP7B. Interestingly, dysregulation of the Cu transport mechanism in the choroid plexus of Atp7b –/– mice has been demonstrated in recent studies; these mice exhibited low Cu levels in the brain at 4 weeks postnatally, emphasizing the crucial role of ATP7B in Cu accumulation46. In summary, the inhibition of CTR1 could regulate the cuproptosis pathway by affecting Cu uptake. It is yet to be established whether the upregulation of ATP7A and ATP7B expression following CI-RI serves as a protective mechanism by promoting Cu efflux or accumulation. However, both ATP7A and ATP7B levels and total Cu ion concentration were increased after CI/RI, which is consistent with the anticancer effect of Cu ionophore. This discrepancy can be partly attributed to the limited understanding of the complex mechanisms of action of ATP7A and ATP7B in the brain (Fig. 9).

The inflammation of nerve cells in cerebral ischemia-reperfusion injury can be mitigated by Disulfiram through the down-regulation of FDX1.
The immune system relies on Cu to execute certain functions. Recent studies have demonstrated that even in the presence of a marginal deficiency, Cu contributes to a decrease in IL concentrations. Neutrophils accumulate Cu as they differentiate into more mature cell populations, and this accumulation is not reflected by an increase in Cu/Zn superoxide dismutase or cytochrome c oxidase activity47 HSP70 is induced by Cu exposure48 TLR4 is a PRR that recognizes specific DAMPs such as HSP70, which, in turn, triggers TLR4-mediated inflammatory responses. Thus, HSP70 in the CSF may function as a neuroinflammatory mediator17. The inflammasome protein complex has recently emerged as a pivotal component of the innate immune response during ischemic stroke49. NLRP3 has been extensively studied in central nervous system diseases50. The activation of the NLRP3 inflammasome necessitates TLR4-mediated activation of the p65 subunit within the downstream NFκB pathway51. Subsequently, NLRP3 binds to ASC to form a complex, and Caspase-1 specifically cleaves GSDMD, releasing the N-terminus from its self-inhibitory C-terminus. GSDMD-N binds to lipids to form non-selective pores, resulting in cell membrane rupture and promoting the release of a large number of inflammatory factors, ultimately triggering pyroptosis52. With the maturation and efflux of IL-18 and IL-1β, Th17 cells and γδT cells release interleukin-17 (IL-17), an important factor in the adaptive immune system53. Several studies have shown that IL-17 is associated with the pathogenesis of cerebral ischemia–reperfusion. Loss of γδT cells alleviates brain tissue damage after ischemia–reperfusion, and IL-17 positive lymphocytes are also detected in brain tissue validated in the field of stroke patients54. The activated NLRP3 inflammasome promotes the activation of IL-17 in CI/RI50.
The anti-inflammatory efficacy of DSF has been substantiated in numerous studies. However, its role in CI-RI remains elusive. In the current study, we found that the association between CI/RI and Cu-induced cell death was accompanied by an enhanced inflammatory response (classic pyroptosis), which was mediated via the HSP70/TLR4/NLRP3 pathway. Importantly, the administration of DSF effectively inhibited this pathway to alleviate CI/RI. Our findings suggest that DSF exhibits robust safety as a well-established pharmaceutical employed in clinical settings for an extensive duration. This finding challenges the conventional belief that DSF, in conjunction with Cu ionophores, forms a complex that effectively treats cancer and induces apoptosis, specifically in cancer cells. However, conventional copper chelators hamper angiogenesis, impeding brain tissue recovery following ischemia. Hence, further investigations into drug applications are warranted. Collectively, the findings of the current study further suggest the presence of potential crosstalk among multiple RCDs in ischemic stroke-induced neuroinflammation, such as pan-apoptosis, potentially mediated via multiple mechanisms.
Conclusions
The CI/RI model in C57BL/6 mice revealed that the cuproptosis-induced inflammatory pathway contributes to CI/RI and that DSF exerts a protective effect on mitochondria while reducing the cerebral infarct size by inhibiting FDX1-mediated protein esterification and HSP70-mediated inflammatory response. The findings of the current study present a novel concept for enhanced mitigation of cerebral blood flow recanalization-induced damage.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Donnan, G. A., Fisher, M., Macleod, M. & Davis, S. M. Stroke. Lancet (Lond., Engl.) 371(9624), 1612–1623 (2008).
Huo, X. & Gaofeng,. Chinese guidelines for endovascular treatment of acute ischemic stroke 2023. Chin. J. Stroke 18(06), 684–711 (2023).
Jurcau, A. & Simion, A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. IJMS 23, 14 (2021).
Chen, D., Guo, Y., Li, X., Zhang, G. & Li, P. Small molecules as modulators of regulated cell death against ischemia/reperfusion injury. Med. Res. Rev. 42, 2067–2101 (2022).
Galluzzi, L. et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 22, 58–73 (2015).
Santagostino, S. F., Assenmacher, C.-A., Tarrant, J. C., Adedeji, A. O. & Radaelli, E. Mechanisms of regulated cell death: Current perspectives. Vet. Pathol. 58, 596–623 (2021).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Tong, X. et al. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol. 15, 174 (2022).
Nie, D., Chen, C., Li, Y. & Zeng, C. Disulfiram, an aldehyde dehydrogenase inhibitor, works as a potent drug against sepsis and cancer via NETosis, pyroptosis, apoptosis, ferroptosis, and cuproptosis. Blood Sci. 4, 152–154 (2022).
Liesz, A. et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 35, 583–598 (2015).
Gadani, S. P., Walsh, J. T., Lukens, J. R. & Kipnis, J. Dealing with danger in the CNS: The response of the immune system to injury. Neuron 87, 47–62 (2015).
Shichita, T. et al. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 23, 723–732 (2017).
Bianchi, M. E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 81, 1–5 (2007).
Land, W. G. Role of heat shock protein 70 in Innate alloimmunity. Front. Immun. 2, 89 (2012).
Li, L., Acioglu, C., Heary, R. F. & Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 91, 740–755 (2021).
Wan, Z. et al. TLR4-HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC-I. Int. Immunopharmacol. 41, 74–81 (2016).
Qu, J. et al. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J. Neuroinflamm. 14, 228 (2017).
Jheng, H.-F. et al. Albumin stimulates renal tubular inflammation through a HSP70-TLR4 axis in early diabetic nephropathy. Dis. Models Mech. https://doi.org/10.1242/dmm.019398 (2015).
Zamora, R. et al. Spatiotemporally specific roles of TLR4, TNF, and IL-17A in murine endotoxin-induced inflammation inferred from analysis of dynamic networks. Mol. Med. 27, 65 (2021).
Kopitar-Jerala, N. Innate immune response in brain, NF-Kappa B signaling and cystatins. Front. Mol. Neurosci. https://doi.org/10.3389/fnmol.2015.00073 (2015).
Ye, Y. et al. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-κB signaling pathway. Front. Cell. Neurosci. 13, 553 (2019).
Franke, M. et al. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 92, 221–231 (2021).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022).
Zhang, M. et al. Association between the change of serum copper and ischemic stroke: A systematic review and meta-analysis. J. Mol. Neurosci. 70, 475–480 (2020).
Guo, Q., Ma, M., Yu, H., Han, Y. & Zhang, D. Dexmedetomidine enables copper homeostasis in cerebral ischemia/reperfusion via ferredoxin 1. Ann. Med. 55(1), 2209735 (2023).
Lu, C., Li, X., Ren, Y. & Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharmacol. 87, 159–172 (2021).
Wright, C. & Moore, R. D. Disulfiram treatment of alcoholism. Am. J. med. 88(6), 647–655 (1990).
Qiu-yang, H., **ao-zhong, C., Dai Chen, Yu., **u-Yan, S. Y. & Zhi-hua, L. Research progress on the role of disulfiram in inflammation-related diseases. West China J. Pharm. 01, 117–122 (2023).
Deng, W. et al. Disulfiram suppresses NLRP3 inflammasome activation to treat peritoneal and gouty inflammation. Free Radic. Biol. Med. 152, 8–17 (2020).
Bai, Y. et al. Disulfiram blocks inflammatory TLR4 signaling by targeting MD-2. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.2306399120 (2023).
Li, H. et al. The combination of disulfiram and copper for cancer treatment. Drug Discov. Today 25(6), 1099–1108 (2020).
Skrott, Z. et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 552(7684), 194–199 (2017).
Lu, Y. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicine via cuproptosis. J. Mater. Chem. B https://doi.org/10.1039/D2TB01150F (2022).
Longa, E. Z., Weinstein, P. R., Carlson, S. & Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20, 84–91 (1989).
Cumings, J. N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain 71, 410–415 (1948).
Rongzhu, L. et al. Zinc, copper, iron, and selenium levels in brain and liver of mice exposed to acrylonitrile. Biol. Trace Elem. Res. 130, 39–47 (2009).
Lutsenko, S., Bhattacharjee, A. & Hubbard, A. L. Copper handling machinery of the brain. Metallomics 2, 596 (2010).
Zhong, C.-C. et al. Copper (Cu) induced changes of lipid metabolism through oxidative stress-mediated autophagy and Nrf2/PPARγ pathways. J. Nutr. Biochem. 100, 108883 (2022).
Zhang, Y. et al. Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients 15(4), 972 (2023).
Lee, J., Petris, M. J. & Thiele, D. J. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. J. Biol. Chem. 277, 40253–40259 (2002).
Tümer, Z. & Møller, L. B. Menkes disease. Eur. J. Hum. Genet. 18, 511–518 (2010).
Lutsenko, S. Dynamic and cell-specific transport networks for intracellular copper ions. J. Cell Sci. https://doi.org/10.1242/jcs.240523 (2021).
Petris, M. J. et al. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 15, 6084–6095 (1996).
Hung, I. H. et al. Biochemical characterization of the Wilson disease protein and functional expression in the yeast saccharomyces cerevisiae. J. Biol. Chem. 272, 21461–21466 (1997).
La Fontaine, S. & Mercer, J. F. B. Trafficking of the copper-ATPases, ATP7A and ATP7B: Role in copper homeostasis. Arch. Biochem. Biophys. 463, 149–167 (2007).
Washington-Hughes, C. L. et al. Atp7b-dependent choroid plexus dysfunction causes transient copper deficit and metabolic changes in the develo** mouse brain. PLoS Genet. 19, e1010558 (2023).
Percival, S. S. Copper and immunity. Am. J. Clin. Nutr. 67(5 Suppl), 1064S-1068S (1998).
Urani, C., Melchioretto, P., Morazzoni, F., Canevali, C. & Camatini, M. Copper and zinc uptake and hsp70 expression in HepG2 cells. Toxicol. In Vitro. Int. J. Publ. Assoc. BIBRA 15(4–5), 497–502 (2001).
Walsh, J. G., Muruve, D. A. & Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 15, 84–97 (2014).
Wang, H. et al. NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 227, 101–113 (2019).
Luo, L. et al. Intermittent theta-burst stimulation improves motor function by inhibiting neuronal pyroptosis and regulating microglial polarization via TLR4/NFκB/NLRP3 signaling pathway in cerebral ischemic mice. J. Neuroinflamm. 19(1), 141 (2022).
Coll, R. C., Schroder, K. & Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 43, 653–668 (2022).
Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517 (2009).
Zhang, J., Mao, X., Zhou, T., Cheng, X. & Lin, Y. IL-17A contributes to brain ischemia reperfusion injury through calpain-TRPC6 pathway in mice. Neuroscience 274, 419–428 (2014).
Funding
This work was supported by the Heilongjiang Province Key R&D Program (Grant no. JD22C002 and GA21C005).
Author information
Authors and Affiliations
Contributions
SY conceived the idea of the study,performed experiments and wrote the manuscript. XL designed the experiments. JY, FJ, XF, JJ and WZ analyzed the data. GL and DZ revised the final manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., Li, X., Yan, J. et al. Disulfiram downregulates ferredoxin 1 to maintain copper homeostasis and inhibit inflammation in cerebral ischemia/reperfusion injury. Sci Rep 14, 15175 (2024). https://doi.org/10.1038/s41598-024-64981-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64981-x
- Springer Nature Limited