
Abstract
Halophiles are one of the classes of extremophilic microorganisms that can flourish in environments with very high salt concentrations. In this study, fifteen bacterial strains isolated from various crop rhizospheric soils of agricultural fields along the Southwest coastline of Saurashtra, Gujarat, and identified by 16S rRNA gene sequencing as Halomonas pacifica, H. stenophila, H. salifodinae, H. binhaiensis, Oceanobacillus oncorhynchi, and Bacillus paralicheniformis were investigated for their potentiality to produce extremozymes and compatible solute. The isolates showed the production of halophilic protease, cellulase, and chitinase enzymes ranging from 6.90 to 35.38, 0.004–0.042, and 0.097–0.550 U ml−1, respectively. The production of ectoine-compatible solute ranged from 0.01 to 3.17 mg l−1. Furthermore, the investigation of the ectoine-compatible solute production at the molecular level by PCR showed the presence of the ectoine synthase gene responsible for its biosynthesis in the isolates. Besides, it also showed the presence of glycine betaine biosynthetic gene betaine aldehyde dehydrogenase in the isolates. The compatible solute production by these isolates may be linked to their ability to produce extremozymes under saline conditions, which could protect them from salt-induced denaturation, potentially enhancing their stability and activity. This correlation warrants further investigation.
Similar content being viewed by others


Introduction
Extremophiles are organisms that can thrive in environments with extreme conditions such as extreme temperature, pressure, radiation, salinity, pH levels, etc.1,2 that have traditionally been considered inhospitable for life3. These organisms cope with harsh environments by adopting certain unique strategies for survival. For instance, the first reported extremophiles termed halophiles are known to have developed two classic strategies called ‘high-salt-in-cytoplasm’ and ‘salt-out-in-cytoplasm’ or ‘low-salt-high-compatible-solute-in-cytoplasm’ for osmoadaptation4. The former mechanism involves the accumulation of intracellular KCl concentrations higher than the external NaCl concentration to maintain the turgor pressure5. However, the resulting high ionic strength in their cytoplasm cost them a compensatory evolutionary change from normal to acidic proteome to keep the proteins soluble for maintaining normal functionality of key cellular activities thereby confining their adaptability to only hypersaline habitats without frequent fluctuations6 to typically 5 M NaCl or more, found mostly in extreme halophiles such as Halobacteriales (archaea), Salinibacter ruber (bacteria) and Haloanaerobiales (anaerobic moderate halophilic bacteria)5,7. Although energetically more expensive, the latter strategy employs a physiologically much more flexible mechanism involving either an accumulation of high concentrations of organic compatible solutes in the cytoplasm from external environments or their de-novo synthesis7 for osmotic adjustments thereby circumventing the long-lasting large-scale accumulation of ions6. This strategy facilitates the adaptability of the organisms possessing them to a wide range of salinities (typically 0.5–3 M NaCl). It is found largely in halotolerant and moderate halophiles5. These halophilic extremophiles also produce a special and unique enzyme called extremozymes to survive in intolerably hostile environments3. These extremozymes are known for their promising capability to withstand unusual extreme conditions required in industrial product synthesis processes where the mesophilic enzymes usually precipitate or denature8. They reportedly replace chemical catalysts in many industries, such as manufacturing chemicals, textiles, pharmaceuticals, detergents, food, paper, etc.9,10. Halophiles, reportedly one of the most important groups of such extremophiles are microorganisms that can flourish in environments with very high salt concentrations. They include members of all three domains of life, viz. Archaea, Bacteria, and Eukarya. In contrast, bacteria that can tolerate relatively high NaCl concentrations and grow regardless of salt's presence or absence are labeled halotolerant11.
Halophilic microorganisms have been reported as an excellent source of extremozyme called halozymes that can function stably under high salt concentrations and withstand high temperatures, alkaline pH, toxicants, etc. encountered in many industrial bioconversion processes12. The polyextremophilic nature of their enzymes makes halophiles a potential candidate for meeting the current industrial enzyme demands. Many scientists have reported their stability and dynamic performance in multiple extreme conditions, such as low water activity environments, aqueous/organic and non-aqueous solvents or media, etc.8,13. Such novel halozymes reported in halophiles include proteases, amylases, lipases, xylanases, nucleases, cellulases, catalases, and esterases8.
The ability of these halophiles to produce beneficial lysis enzymes such as chitinase, cellulase, and protease implies their direct potential use as biocontrol agents for controlling phytopathogenic fungi, pests, nematodes and for rhizospheric soil decomposition for increasing plant nutrient availability for sustainable agriculture and as livestock feed additives, etc.14. Besides, they also find applications in many industries such as food and feed, laundry and detergent, leather and textiles, pulp and paper, alcohol and beverages, medicine and pharmaceuticals, environmental bioremediations, biomass conversions for biofuel production, etc. So, given their profound agricultural and industrial significance, these enzymes viz., chitinase, cellulase, and protease were chosen in this study to unlock their production potential within the halophilic and halotolerant bacterial isolates to unveil and shed light on their vast potential applications.
These industrial enzymes are found in various sources, such as plants, animals, and microorganisms. However, microbial sources are preferred due to their high stability, cost-effectiveness, less time and space requirement, high consistency, production and optimization ease, and increasing demand in many industries15, as evidenced by their total contribution of more than 82% of revenue share in 202216. However, despite these enzymes being widely studied in many organisms, only a few reports have been made on the extracellular enzymes of halophilic and halotolerant bacteria, especially Halomonas sp.17. Besides, halophiles have been isolated and investigated for many other possible biotechnological applications, such as the production of compatible solutes, enhanced oil recovery, and the degradation of industrial pollutants in saline habitats and as potential agricultural bioinoculants for the recovery of saline soils11,18.
Compatible solutes or osmoprotectants are low molecular weight organic molecules with a neutral charge and low toxicity at high concentrations either accumulated from external environments or secreted by halophiles in their cytoplasm to act as osmolytes for their survivability against the extreme osmotic stresses19. They include several different classes, such as high water-soluble sugars, alcohols or polyols, betaines, amino acids, ectoine, and its derivatives, among which ectoine and glycine-betaine are reported as the most predominant ones. They are used in many biotech industries for stabilizing enzymes, DNA, and whole cells against freezing and thawing, drying and heating, and denaturants such as urea and salts, and as salt antagonists, stress-protective agents, moisturizers, therapeutics, and for increasing the freshness of foods in food industries20,21. In plants, their accumulation is said to increase survivability against various stresses such as salinity, heat, and drought. The genetic manipulation of these osmoprotectants' responsive genes has been suggested as one of the strategies to improve plant stress tolerance by enhancing their production22,23,24.
In light of the above perspectives, the current research was conducted to study the production potentiality of extracellular halozymes viz. protease, cellulase, and chitinase, and ectoine compatible solute, and PCR based molecular detection of the biosynthetic gene of ectoine and glycine betaine of halophilic and halotolerant bacteria isolated from the crop rhizospheric soils of agricultural fields of southwest coastline of Saurashtra Gujarat.
Results
Preliminary soil analysis
The preliminary soil analysis results are summarized in Table 1. The physicochemical properties of the soil samples, including pH, electrical conductivity (E.C.), organic carbon content, and availability of phosphorous and potash ranged from 7.4 to 8.1, 0.76–1.59 dS m−1, 4.03–7.47 g kg−1, 29.57–54.33 kg ha−1 and 166.70–248.33 kg ha−1 respectively11.
Halophilic characterization
The characterization of the isolates by NaCl tolerance test showed that the isolates could tolerate up to 25% with optimum growth between 10 and 15% NaCl concentrations11. The isolates S1 through S9 and S11, all belonging to the Halomonas species, exhibited remarkable and robust growth despite challenging salt concentrations exceeding 10–15% NaCl. These Halomonas isolates surpassed their counterparts S10, S12, S13, S14, and S15 belonging to the Oceanobacillus and Bacillus species with exceptional vigor. Intriguingly, the Halomonas species isolates maintained a consistently thriving growth pattern even at higher NaCl concentrations, with only a modest decline observed up to 25% NaCl. This remarkable resilience and adaptability demonstrated by the Halomonas isolates highlight their exceptional ability to thrive in extremely saline environments. In stark contrast, the Oceanobacillus and Bacillus species isolates, S10, S12, S13, S14, and S15, exhibited a stark decline in their growth trajectory, indicating their limited capacity for salt tolerance and a considerably less robust response to the challenging conditions presented by 25% NaCl concentration.
Microscopic characterization
The gram staining analysis of the isolates revealed that all the Halomonas species isolates exhibited gram-negative characteristics, while the Bacillus species isolates exhibited gram-positive characteristics at the microscopic level. This result was complemented by the identification of isolates based on 16S rRNA partial gene sequencing. The scanning electron microscopic characterization identified the isolates as short to thin rod-shaped bacteria in single or pairs to bunchy type organization (Fig. 1) with sizes ranging from 0.38 to 0.83 μm by 0.75–6.78 μm11. All the isolates were motile, as observed under the microscope.

Microscopic characterization of isolates by Scanning Electron Microscope (SEM).
Molecular identification and phylogenetic analysis
The analysis of the partial 16S rRNA gene sequence of the isolates to determine their genetic relatedness and taxonomic identity by comparing their sequences to the known reference strains in the curated 16S database of EzBioCloud revealed that isolates S1, S3, S5, S6, S8, and S11 belonged to Halomonas pacifica with percent similarity of 99.15%, 99.45%, 99.52%, 99.45%, 99.01% and 99.52% with H. pacifica NBRC 102220 respectively (Table 2). In contrast, isolate S2 belonged to Halomonas stenophila with a percent similarity of 99.12% with H. stenophila N12. Isolates S4 and S7 belonged to H. salifodinae with percent similarity of 99.34% and 99.22% respectively while isolate S9 belonged to Halomonas binhaiensis with 99.52% similarity with Halomonas binhaiensis Y2R2 (Table 2). On the other hand, isolate S10 belonged to Oceanobacillus oncorhynchi with a percent similarity of 98.23% with Oceanobacillus oncorhynchi subsp. oncorhynchi R-2 while isolates S12, S13, S14 and S15 belonged to Bacillus paralicheniformis with percent similarity of 99.59%, 99.46%, 98.19% and 98.44% with B. paralicheniformis KJ-16 (Table 2). The partial 16S rRNA gene sequence of isolates S1 and S2 were submitted to NCBI with accession numbers MK955347 and MK961217 re-designating the isolates as Halomonas pacifica HPSB1 and Halomonas stenophila HPSB2 respectively11. The BLAST analysis demonstrated the highest level of genetic relations between the respective Halomonas, Oceanobacillus, and Bacillus isolates which features and supports their taxonomic uniformity within the genus. The phylogenetic analysis of these isolates, along with their genetically closest reference species, constructed using MEGA11 following the Minimum Evolution tree method using their multiple sequence alignment (MSA) aligned by ClustalW, and analyzed based on the maximum composite likelihood substitution model, distinctly elucidated the interconnectedness among various genera within their respective species, as visually depicted in Fig. 2.

Phylogenetic dendogram, based on 16S rRNA nucleotide sequences, showing the genetic interrelationship among the different halophlic and halotolerant bacterial isolates within the closely related species of various genera.
Quantification of protease, cellulase, and chitinase enzymes
The results of the quantitative protease, cellulase, and chitinase enzyme assays and the respective enzymes’ specific activities of the isolates are presented in Supplementary Tables 1, 2, and 3, respectively. The various standard curves viz. tyrosine, glucose, NAG, and BSA used to estimate the protease, cellulase, and chitinase enzymes, and protein content of the isolates in the respective enzyme production medium are shown in Supplementary Figs. 1, 2, 3, and 4 respectively.
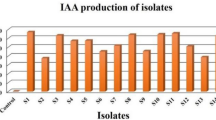
The protease, cellulase, and chitinase activities of the isolates ranged from 6.90 to 35.38 U ml−1 min−1, 0.004–0.042 U ml−1 min−1, and 0.097–0.550 U ml−1 h−1 respectively, while their respective corresponding specific activities ranged from 7.23 to 36.21 U mg−1 min−1, 0.007–0.062 U mg−1 min−1, and 0.146–0.471 U mg−1 h−1, respectively. The highest protease, cellulase, and chitinase activities were shown by isolate S13, while the lowest was shown by isolates S4 (Fig. 3), S5 (Fig. 4), and S7 (Fig. 5), respectively. On the other hand, the highest protease, cellulase, and chitinase-specific activities were shown by isolates S15 (Fig. 3), S12 (Fig. 4), and S13 (Fig. 5), respectively while the lowest was shown by isolates S4, S5, and S7, respectively.

Protease activity of halophilic and halotolerant bacterial isolates (U ml−1 min−1).

Cellulase activity of halophilic and halotolerant bacterial isolates (U ml−1 min−1).

Chitinase and its specific activity and protein content of halophilic and halotolerant bacterial isolates.
The protein content of the isolates in their respective protease, cellulase, and chitinase enzyme production medium ranged from 0.80 to 1.28 mg ml−1, 0.450–0.908 mg ml−1, and 0.550 to 1.166 mg ml−1, respectively. Isolate S13 exhibited the highest protein content in the protease, cellulase, and chitinase enzyme production medium, while isolates S3 (Fig. 3), S2 (Fig. 4), and S3 (Fig. 5), respectively, showed the lowest protein content. The statistical analysis of variance (ANOVA) for all the data of tyrosine, glucose, and NAG released, protease, cellulase, and chitinase activity, protein contents, and respective enzymes’ corresponding specific activities of the isolates indicated a high level of significance inferred from the greater value of calculated F than that of table F at both the 1% and 5% significance levels.
Ectoine production potentiality
The results of the ectoine production potentiality of isolates are presented in Table 3. The ectoine production of the isolates was determined according to the peak generated by the ectoine standard at varying concentrations in LCMS (Fig. 6). The ectoine production ranged from 0.01 to 3.17 mgl−1 shown by the isolates S9 and S10 and S5, respectively (Fig. 7). The chromatogram and mass spectrum profile of the highest ectoine production by the isolate S5 is shown in Fig. 8. However, five out of fifteen isolates showed no detectable ectoine production, perhaps due to deficient ectoine production below the detection threshold limit as confirmed by the presence of their ectoine biosynthetic gene described below in PCR based molecular detection of ectoine biosynthetic gene.

(a) Chromatogram, and (b) mass spectrum profile of ectoine standard.

Production profile of ectoine by different isolates.

(a) Chromatogram, and (b) mass spectrum profile of highest ecoine production by isolate S5.
PCR amplification of ectoine and glycine betaine biosynthetic genes
The PCR amplification targeting the ectC and BADH1 genes confirmed their presence in all fifteen isolates, yielding amplicons of 370 bp (Fig. 9) and 1473 bp (Fig. 10), closely aligning with the sizes reported by Rajan et al.25 for ectC and Anburajan et al.26 for BADH1. The PCR-based molecular detection of the ectC gene thus confirmed and validated the positive ectoine production result obtained by QTOF LCMS as described above.

PCR amplification of ectC gene in halophilic and halotolerant bacterial isolates. L: 100 bp DNA ladder; NTC: no template control; 1–15: bacterial isolates.

PCR amplification of BADH1 gene in halophilic and halotolerant bacterial isolates. L: 1 kb DNA ladder; NTC: no template control; 1–15: bacterial isolates.
Discussion
The NaCl tolerance test revealed that the isolates were moderate halophiles and halotolerant in nature based on the concentration of salt required for optimum growth and their maximum salt tolerance capacity as per the most widely accepted classification by Kushner and Kamekura11,27. This finding was further confirmed and validated by the molecular identification of the isolates by 16S rRNA gene sequencing at genus and species levels. The 16S rRNA gene sequence of the isolates belonging to H. pacifica and H. stenophila were submitted to NCBI with accession numbers MK955347 and MK961217, respectively11.
The protease activity of the isolates belonging to Halomonas species was approximately twofold higher than that of H. meridian HC4321C1 reported by Anithajothi et al.28. For those isolates belonging to Bacillus and Oceanobacillus species, the protease activity was in line with that of B. licheniformis P003 and Oceanobacillus aidingensis, reported by Sarker et al.29 and Kumar et al.13, respectively. However, the findings of many reports suggest that B. licheniformis strains are capable of producing much higher protease activity ranging above 100 to more than a few 1000 U ml−130,31,32 depending upon the degree of culture enrichment with different sources of nitrogen, carbon, and substrates, etc., and is supposedly reported as one of the industrial strain of choice for enzyme production. The protease activity of H. pacifica and H. stenophila is being reported for the first time in our study. The optimum enzyme production potentiality of any microorganism is said to depend on the degree of optimized conditions for many factors such as pH, temperature, incubation period, agitation rate, substrate type, sources of carbon, nitrogen, etc., therefore, subject to vary from genus to genus and species to species. These results thus suggest that the isolates under study present suitable candidates for producing protease enzymes for various biotechnological and industrial applications.
The cellulase activity of the isolates belonging to Halomonas species was found to agree with the report of Shivanand et al.12 but much less than that of Halomonas sp. PV1 reported by Benit et al.33. In contrast, the isolate belonging to Bacillus paralicheniformis showed slightly less than the lowest cellulase activity of the same species reported by da Silva et al.34. However, the cellulase activity of the isolates belonging to Bacillus species was at least tenfold higher than that of isolates belonging to Halomonas species. These findings imply that Bacillus species have higher cellulase production potential than those Halomonas species. On the other hand, the cellulase activity of the isolate belonging to Oceanobacillus oncorhynchi was nearly tenfold lesser than that of Oceanobacillus profundus reported by Gbenro et al.35. Nevertheless, the ability of the isolates to produce cellulase enzyme suggests their ability to degrade cellulose, thereby implying the need for further investigation to uncover their fullest potential for the production of the same on a larger scale by providing the best optimum production conditions and suitable infrastructure facilities.
The highest chitinase activity shown by the isolate belonging to B. paralicheniformis was found to agree with the report of Akhir et al.36. While it was approximately tenfold higher than that of B. licheniformis JP2 reported by Keliat et al.37, Hussin and Majid38 have reported even a much lesser chitinase activity of similar species. However, the highest chitinase activity observed in our isolates is still much lesser than that of similar species reported by scientists such as Akeed et al.39 and Sasi et al.40. On the other hand, the chitinase activity of halophilic bacterial species is very limited to date, specifically of Halomonas species, and is being reported for the first time in our study. Furthermore, few earlier reports on the chitinase activity of halophilic bacteria such as Virgibacillus marismortui M3-2341 and Planococcus rifitoensis M2-2642 do prove the chitinase production potentiality of halophilic bacteria. However, there are multiple reports on the chitinase activity of many halotolerant bacteria, particularly that of Bacillus species which is supposedly used as one of the commercial, industrial strains. Nonetheless, the ability of our isolates to produce chitinase enzyme does suggest their ability to degrade chitin compounds, thereby representing a potential biocontrol agent for sustainable agriculture besides various other applications in many industries.
The ectoine production by all the isolates was found to be negligible to significantly less as compared to that of different Halomonas species reported by Zhang et al.43, Van-Thuoc et al.44, Van-Thuoc et al.45, Chen et al.46, Chen et al.47 where ectoine production ranged from 3.65 to 13.96 g l−1. The ectoine production of the isolates belonging to H. pacifica, H. stenophila, H. salifodinae, H. binhaiensis, O. oncorhynchi, and B. paralicheniformis has not been reported earlier. It is being reported for the first time in our study.
The lower amount of ectoine production by the isolates may be attributed to the unoptimized rate of agitation, which is reported to impact oxygen transfer in which a higher agitation rate for some halophiles results in higher dissolved oxygen (DO) level and thereby higher ectoine production. In contrast, a higher agitation rate has also been reported to result in a high shearing force of agitation, lowering microbial growth and ectoine production at an agitation rate higher than 200 rpm47. Furthermore, Chen et al.47 achieved higher ectoine production potentiality after utilizing a well-optimized production system with the best carbon and nitrogen sources, optimum ratio, optimum NaCl concentration, and agitation rate. Nevertheless, the production of ectoine by the isolates, even though in smaller quantity, still implies their potentiality for the production of ectoine-compatible solute.
The ectC gene amplified in the isolates is reported to encode putative proteins of 129 amino acids and codes for the L-ectoine synthase protein that catalyzes the final step of the ectoine biosynthetic pathway leading to the synthesis of ectoine osmolyte25. On the other hand, the Betaine Aldehyde Dehydrogenase gene is reportedly encoded by a polynucleotide of 1473 bp (Fig. 10) and polypeptides of 490 amino acids26. The BADH1 gene catalyzes the conversion of betaine aldehyde to glycine betaine in the last step of the biosynthetic pathway that leads to the synthesis of the effective compatible solute glycine betaine, which maintains the fluidity of membranes and protects the biological structure of the organisms under salt stress conditions26. Additionally, similar to ectoine, glycine betaine also aids in stabilizing key proteins like proteases, cellulases, and chitinases against salt-induced denaturation, which are essential for nutrient acquisition and energy metabolism from organic substrates in extreme environments. The presence of the BADH1 gene in the halophilic and halotolerant bacterial isolates suggests their potential to produce glycine betaine, expanding their repertoire of compatible solutes beyond ectoine thereby enabling osmolyte switch. This finding thus emphasizes the diverse adaptive strategies employed by halophilic and halotolerant bacteria to thrive in saline environments, aligning with previous reports of glycine betaine production in related halotolerant species such as B. halodurans SMBPL0626 and B. subtilis MA0448 thereby implying its importance as one of the reportedly most predominant solutes produced besides ectoine in true halophiles studied till date49.
The ability of ectoine-compatible solute production by halophilic bacteria belonging to Halomonas species is evidenced by comprehensive reports on ectoine production in many other Halomonas species, such as H. elongate50,51,52, H. boliviensis53 and halotolerant bacteria such as Bacillus halodurans25. Likewise, the production of glycine betaine has also been reported in halotolerant bacteria such as B. halodurans26, B. subtilis48, etc. The accumulation of these compatible solutes has been reported to confer osmotolerance in plants. The hyperosmotic tolerance conferred by the genetic transformation of the ectoine biosynthetic gene has already been reported in many plants, such as tobacco54,55 and tomato plants56. Similarly, the salt tolerance conferred by transforming the glycine betaine biosynthetic gene has been reported in barley57 and wheat58. The ability of the isolates to produce these compatible solutes thus showed their significance as a source of osmoprotectant responsive genes, which hold a tremendous potentiality for conferring osmotolerance to plants through their genetic transformations.
The application of compatible solutes in various industries for stabilizing enzymes suggests a possible correlation that the ectoine-compatible solute produced by the isolates could be involved in aiding the production of extremozymes under saline conditions by preventing their denaturation from salinity and thereby maintaining their production. Enzymes like protease, cellulase, chitinase, etc. are produced and secreted by halophiles to acquire nutrients and energy from organic substrates present in their extreme environments. However, the high salt concentrations in their environments can disrupt the structure and function of these enzymes by interfering with their electrostatic interactions and hydrogen bonding. So, to counteract these denaturing effects of salt, halophiles have evolved to produce these compatible solutes not only as part of their adaptation to saline environments but also to protect and stabilize their metabolically and physiologically important enzymes for survivability. These compatible solutes are said to form protective hydration shells around the enzymes, shielding them from the disruptive effects of salt ions. This hydration stabilizes the enzyme's structure and allows it to remain active and functional in the presence of high salt. Notably, the observations made by Roberts20 and Detkova et al.21 affirm this correlation between compatible solutes and extremozymes production. They documented the multifaceted role of compatible solutes in halophilic bacteria, emphasizing not only their pivotal function in osmoregulation to maintain cellular osmotic equilibrium but also their significant capacity to serve as effective stabilizers of proteins and even whole cells. These findings collectively underscore the intimate connection between compatible solute accumulation and the production and stability of extremozymes, including proteases, cellulases, and chitinases, in halophilic microorganisms.
Materials and methods
The investigation was carried out at the “Department of Biotechnology, College of Agriculture, Junagadh Agricultural University, Junagadh” during 2019–2022.
Isolation of bacteria
The halophilic and halotolerant bacteria were isolated from 15 different soil samples, each approximately 100 g in weight, collected from various crop rhizospheres in different agricultural fields lying along the southwest coastline of Saurashtra, Gujarat. Specifically, samples were obtained from Junagadh and Porbandar districts, located at coordinates 21.52° N 70.47° E and 21°37′48″ N 69°36′0″ E, respectively, as outlined in Table 4, as previously reported by Reang et al.11. Following the streak plate method, the bacteria were isolated from 10 ml of soil suspensions (prepared from 1 g) by streaking a loopful of the 10–5 dilution onto a freshly prepared autoclaved halophilic agar media supplemented with 10% NaCl, adjusted to pH 7.2 ± 0.2 (at 25 °C), and incubated at 37 °C for 5 days. The isolates were characterized for halophilic and halotolerant nature by subjecting them to a varying concentration of NaCl ranging from 5, 10, 15, 20, and 25% in halophilic broth for salt tolerance test. Pure culture plates of the above isolates were prepared on the same media and used to prepare primary inoculum seed culture.
Preliminary soil analysis
The preliminary analysis of soil samples was conducted to assess soil chemical properties, including soil pH using potentiometry and electrical conductivity via the conductometry method59. Soil organic carbon content was determined using the back titration method60, while available soil phosphorus was measured using a colorimetric method61, and soil potash was analyzed via flame photometry59.
Preparation of inoculum
A primary inoculum of the isolates was prepared by inoculating a single colony from each pure culture plate as prepared above on a freshly prepared autoclaved 10 ml halophilic broth in test tubes and incubated at 37 °C for 24 h. As described below, the primary inoculum was then used as seed culture for the extracellular enzymes and compatible solutes production potentiality experiments.
Microscopic characterization of isolates
Gram’s staining
A thick smear of all cultures was prepared on a clean glass slide individually and subjected to the gram staining process. Subsequently, the slides were allowed to air dry at room temperature overnight and observed under the Zeiss Imager.Z2 optical microscope to ascertain the orientation of the isolates.
Scanning electron microscopy
A loopful of the bacterial isolates colony was picked from their respective fresh culture plates, and a light smear was made on the aluminium stub with the help of inoculating needle. The smeared stub was then flooded with 4% glutaraldehyde and kept in a fridge at 4 ℃ for 24 h. The following day the smeared samples were dehydrated by using a gradient dilution of acetone in a concentration ranging from 30, 50, 70, 80, 90, and 100% and treating each sample by dip** into dilution of each respective concentration in the order of 30–100% for 15 min. The samples treated by dip** in an acetone concentration of 100% were repeated for a second time for another 15 min for each sample. The dehydrated samples were then coated in a spotter coater with a gold–palladium mixture plate and observed under a scanning electron microscope (Zeiss EVO 18).
Motility test
The bacterial motility test was done by the hanging drop method. A few drops of liquid culture were placed onto the coverslip sterilely. A depression slide was taken, and the concave portion over the drop pressed the slide onto the cover slip. The slide was inverted quickly to keep from disrupting the drop. Then the motility was examined under the Zeiss Imager.Z2 optical microscope at 40× magnification.
Molecular identification of bacterial isolates
The identification of halophilic and halotolerant bacterial isolates was performed using BLAST analysis of their partial 16S rRNA gene sequences against known reference strains in the curated 16S database of EzBioCloud. Reference strains exhibiting a sequence similarity threshold above 96% with the query isolates were selected for further analysis. A phylogenetic tree was constructed using MEGA11, applying the maximum composite likelihood substitution model with 100 bootstrap replications. The tree was generated under the minimum evolution method to elucidate the genetic interconnectedness among various genera within their respective species.
Protease enzyme production
The protease enzyme production was carried out by inoculating 1% of the test isolates in a 250 ml Erlenmeyer flask containing 100 ml autoclaved protease production broth prepared by dissolving 3% nutrient gelatin, 0.8% nutrient broth, 0.5% casein, 0.01% MnCl2, 15% NaCl, and 1.2 ml of 20% glycerol and incubated at 37 ℃ for 72 h in a shaker incubator (150 rpm). After 72 h of growth, the cells were harvested at 10,000 rpm for 15 min, and the supernatant thus obtained was used as crude enzyme for quantitative assay.
Protease enzyme quantification assay
The protease enzyme assay followed Sigma's non-specific protease assay described by Cupp-Enyard62. The assay was performed in triplicates in 15 ml test tubes using enzyme extracts from each isolate, with one tube serving as a blank control. In each set of four tubes, 5 ml of 0.65% casein solution was added and equilibrated at 37 °C for 5 min. Then, 1 ml of enzyme extract was introduced to three of the tubes (excluding the blank), mixed, and incubated at 37 °C for 10 min. Tyrosine release and protease activity were measured and compared among test isolates during this incubation period. After 10 min, 5 ml of 110 mM TCA reagent was added to stop the reaction and incubated for 30 min at 37 °C. Concurrently, a series of standard tyrosine dilutions were prepared in six test tubes, with incremental volumes of 1.1 mM tyrosine standard stock solution, precisely measuring 0.00, 0.05, 0.10, 0.20, 0.40, and 0.50 ml, respectively. Subsequently, each standard dilution was adjusted to a final volume of 2 ml by the addition of an appropriate volume of purified water.
After a 30-min incubation, each test solution and blank was filtered using a 0.45 μm polyethersulfone syringe filter to eliminate insoluble components. The resulting 2 ml filtrate from both the test isolates and blanks was transferred to new test tubes. In all the test tubes containing standards and standard blank, 5 ml of sodium carbonate was added, followed immediately by 1 ml of Folin’s reagent to stabilize pH. After observing a cloudy appearance due to the reaction with free tyrosine, the tubes were gently mixed and incubated at 37 °C for 30 min. After incubation, the tubes with standards were seen with a gradation of color correlating with the amount of tyrosine added. This color change was also observed in tubes with test samples. Subsequently, 2 ml of these solutions were filtered into cuvettes using a 0.45 μm polyethersulfone syringe filter. The absorbance of the standards, standard blank, the different test isolates, and test blank were measured in a spectrophotometer at 660 nm wavelength with a 1 cm light path. The standard tyrosine curve was then constructed to determine the amount of tyrosine released and estimate the protease activity of the test isolates using the below-described formula.
One unit of protease activity (U) was defined as the amount of enzyme capable of releasing 1.0 μ mol of tyrosine per min from the casein substrate under the described reaction conditions. The protein content was estimated by Lowry’s method, with bovine serum albumin (BSA) as the standard. The protease enzyme and specific activity were then determined by calculating using the following formula:
where, 11 = Total volume of assay (ml). 10 = Time of assay (min) as per the unit definition. 1 = Volume of enzyme used (ml). 2 = Volume taken in cuvette for colorimetric determination.
Cellulase enzyme production
The cellulase enzyme production was carried out by inoculating 1% of the bacterial isolates in a 250 ml Erlenmeyer flask containing 100 ml autoclaved cellulase production broth prepared by dissolving 1% CMC, 0.2% NaNO3, 0.05% MgSO4, 0.005% K2HPO4, 0.001% FeSO4, 0.002% CaCl2 and MnSO4, 15% NaCl, and incubated on water bath shaker at 120 rpm at 37 °C for 5 days for cellulase enzyme production. After incubation, the bacterial cultures were harvested by centrifugation at 5000 rpm for 20 min. The culture supernatants were used for the quantification of the cellulase enzyme.
Cellulase enzyme quantification assay
The cellulase activity was assayed using the DNSA method, followed by Lay Mg Mg et al.63. One milliliter of culture supernatant was mixed with 1 ml of 0.05 M citrate buffer (pH 4.8) solution in test tubes containing 1% cellulose substrate. The resulting reaction mixture was incubated at 50 °C for 60 min in a water bath shaker at 80–85 rpm. After the reaction time, 3 ml of DNSA reagent was added to the reaction mixture and boiled for exactly 5 min to terminate the reaction in a vigorously boiling water bath. The reaction mixture was then cooled in a cold water bath, and the absorbance was measured by a spectrophotometer at 540 nm against the blank without enzyme filtrate. Anhydrous glucose was the standard64. One unit of cellulase activity (U) was defined as the amount of enzyme capable of releasing 1.0 mg of glucose per min from the cellulose substrate under the described reaction conditions. The protein content was estimated as described above. The cellulase enzyme activity and specific activity were then determined by calculating using the following formula:
where, 5 = Total volume of assay (ml). 0.5 = Dilution factor (DF). DF = (Volm of enzyme extract/Volm of enzyme + buffer). 1 = Volume of enzyme extract used (ml). 2 = Volume of reaction mixture taken in cuvette (ml). 60 = Incubation time (min)
Chitinase enzyme production
The chitinase enzyme production was performed by modifying the standard method followed by Hsu and Lockwood65. One milliliter of the bacterial isolates was inoculated in the 250 ml Erlenmeyer flask containing 100 ml autoclaved Minimal Medium (Designated as MM) broth prepared by dissolving 0.5% colloidal chitin, 0.05% MgSO4∙7H2O, 0.03% KH2PO4, 0.07% K2HPO4, 0.0001% MnCl2, 0.001% FeSO4∙7H2O and 0.0001% ZnSO4, 15% NaCl in 1000 ml distilled water and the final pH was adjusted to 7. The inoculated tubes were incubated in a shaker incubator (200 rpm) at 30 °C for 48 h. After incubation, the isolates' cultures were harvested and used for carrying out further quantitative chitinase enzyme assay. An uninoculated test tube containing the same liquid broth was kept as blank.
Chitinase enzyme quantification assay
Chitinase activity was determined by modifying a colorimetric method followed by Setia and Sohorjono66 in triplicates. The reaction mixture consisted of 1 ml of the crude enzyme and 2 ml of 1.25% (w/v) colloidal chitin substrate in a 200 mM potassium phosphate buffer (pH 6.0). The mixture was incubated at 30 °C for 2 h and boiled for 10 min to stop the reaction, then cooled to room temperature in a cold water bath, and 1 unit of β-N-Acetylglucosaminidase (NAGase) was added and then centrifuged at 8000 rpm for 20 min. The 1 ml of test supernatant obtained from the above centrifugation was added to 1.5 ml of freshly prepared color reagent solution prepared by mixing 96 mM DNSA (3,5-Dinitrosalicylic Acid) reagent in 5.3 M sodium potassium tartrate solution and diluted to 40 ml with deionized water. The test supernatant and color reagent solution mixture was then boiled for 5 min and cooled to room temperature. The concentration of GlcNAc (N-acetylglucosamine) released was then measured at 540 nm. The standard curve of GlcNAc was plotted between GlcNAc concentration and GlcNAc absorbance. One unit of chitinase enzyme activity (U) was defined as the amount of enzyme capable of liberating 1.0 mg GlcNAc per hour from the chitin substrate under reaction conditions.
The protein content in isolates was determined by the Folin-Lowry method using BSA as standard. Data on chitinase enzyme activity, specific activity, and protein content was analyzed with a single factorial CRD analysis of variance (α = 0.05).
The milligrams of NAG liberated were determined using the standard curve, and the chitinase enzyme activity (U ml−1 h−1) and its specific activity (U mg−1 h−1) defined per mg of protein estimated in isolates were then calculated using the following formula:
where, 3 = Initial reaction volume of assay. 2 = Conversion factor for converting 2 h to 1 h as per the unit definition. 1 = Volume (ml) of supernatant used in colorimetric determination. 1 = Volume (ml) of crude enzyme used. Volume of NAGase = 0.5 ml.
Ectoine production potentiality
Culture medium
The ectoine production of the bacterial isolates was carried out in an autoclaved freshly prepared culture medium consisting of yeast extract (86 gl−1), ammonium sulfate (28 gl−1), FeCl2.4H2O (0.5 mM), MnSO4∙H2O (10 μM), KCl (2 gl−1), MgSO4.7H2O (100 mM), and sodium chloride (1 M) as per the modified method of Chen et al.47.
Growth conditions
A primary inoculum seed culture of all the isolates was first prepared by inoculating a loopful of the respective halophilic bacterial cells to 10 ml of autoclaved freshly prepared halophilic broth and incubated in a shaker incubator at 37 ℃ and 180 rpm for 24 h. After 24 h cultivation, 1 ml of the seed culture was inoculated into a 250 ml Erlenmeyer flask containing 50 ml autoclaved freshly prepared ectoine production broth and incubated at 30 ℃ and 200 rpm for 24 h. The flask uninoculated with isolate culture served as the negative control. After cultivation, the samples were taken to analyze cell growth and ectoine concentration.
Ectoine quantification assay
The halophilic bacterial isolates culture cultivated above was harvested by centrifugation at 8000 rpm. The pellets were resuspended in 80% ethanol (v/v) (Sigma) with vigorous shaking for 30 min. The ethanol extracts were filtrated through a 0.45 mm filter to analyze ectoine production. The ectoine concentration was then determined from the filtrate obtained by LCMS as per the details in Table 5. Ectoine, purchased from Sigma, was used as the reference standard.
PCR-based molecular detection of ectoine and glycine betaine biosynthetic genes
The isolates' production potentiality of the ectoine-compatible solute was confirmed by PCR-based molecular detection of its biosynthetic gene ectC which encodes for the ectoine synthase enzyme. Besides, the PCR-based molecular screening also showed the presence of BADH1 gene encoding for betaine aldehyde dehydrogenase enzyme which is responsible for the biosynthesis of glycine betaine compatible solute. The primer sequences for PCR amplification of these genes were obtained from literature reported by Rajan et al.25 and Anburajan et al. 26 for ectoine and glycine betaine, respectively. The isolates' genomic DNA was isolated by Qiagen's blood and tissue kit based on the manufacturer's instructions. The isolated genomic DNA was then analyzed on 0.8% agarose gel electrophoresis, quantified by nanodrop spectrophotometer, and used for further PCR reactions. The PCR reaction was carried out in an Applied Biosystems thermal cycler in a 20 ml reaction volume system containing 50 ng of genomic DNA, 0.5 mM of each primer, 100 mM of each dNTP, 5 U of KAPA Taq DNA polymerase, and 10X Taq A buffer supplemented with 25 mM MgCl2. A single PCR vial containing all the above PCR reaction mixture except the DNA was used as the no template control (NTC) to check the chances of amplification due to primer contamination with the template DNA. The thermal cycler amplification reaction conditions were set with an initial denaturation at 94 ℃ for 5 min, followed by 35 cycles of denaturation at 94 ℃ for 45 s, annealing temperature at 50 ℃ for 45 s, and primary extension at 72 ℃ for 1 min, followed by a final extension at 72 ℃ for 7 min and hold at 4 ℃. The PCR amplified products were checked by running on agarose gel electrophoresis at 90 V cm−1 in 1.5% low EEO agarose gel prepared in 1× TAE buffer and added with 3% of 1000 ppm ethidium bromide. The resulting electrophoresed amplicons of the respective genes were then scanned and captured by the gel documentation system.
Statistical analysis
The above experiments were carried out in triplicate replications. The data obtained from their mean values were used for statistical analysis of variance (ANOVA) using a Completely Randomized Design (CRD) for the interpretation of results.
Conclusion
In the present study, it was thus concluded that the halophilic and halotolerant bacteria isolated from the soils of agricultural fields lying along the southwest coastline of Saurashtra, Gujarat, showed a promising potentiality for production of the industrially important proteolytic, cellulolytic, and chitinolytic extremozymes and may have potential application in many industries especially cellulose and chitinous biomass conversion for biofuel production, etc. Besides, the isolates also represent a potent source of biocontrol agents. They may contribute to sustainable agriculture as an alternative to chemical pesticides against the control and management of fungal diseases, insect pests, and nematodes. However, the isolated halophilic and halotolerant bacteria's reported activities only hint at the possibility of such novel applications. Further study on the candidate isolates’ improvement for higher extremozyme production, their extraction, purification, characterization, and application trials are required for a detailed evaluation of their practical applications. Furthermore, the isolates also exhibited an appreciable potential for producing ectoine-compatible solute, as validated by the presence of its biosynthetic gene. Besides, the isolates also showed the presence of glycine betaine-compatible solute biosynthetic gene. Thus, these isolates may serve as a promising source of osmoprotectant-responsive genes for develo** osmotolerant transgenic plants against salinity, heat, and drought stresses, as already reported by many scientists. The study also suggests that there may be a correlation between compatible solutes and extremozyme production of the isolates under saline conditions. The compatible solutes could be playing a vital role in aiding the maintenance of normal extremozyme production by protecting them from salt-induced denaturation effects, potentially enhancing their stability and activity. However, this hypothesis is purely our assumption, and further investigation is required to confirm it.
Data availability
The main author hereby declares with the consent of all concerned co-authors that data and materials related to the work described would only be made available at request to the corresponding author.
References
Merino, N. et al. Living at the Extremes: Extremophiles and the limits of life in a planetary context. Front. Microbiol. 10, 780 (2019).
Rothschild, L. & Mancinelli, R. Life in extreme environments. Nature 409(6823), 1092–1101 (2001).
Rampelotto, P. H. Extremophiles and extreme environments. Life 3, 482–485 (2013).
Mesbah, N. M., Hanelt, I., Zhao, B. & Muller, V. Microbial adaptation to saline environments: Lessons from the genomes of Natranaerobius thermophilus and Halobacillus halophilus. In Halophiles: Genetics and Genomes (eds. Papke, R. T. & Oren, A.) (Caister Academic Press, 2013)
Patel, S. & Saraf, M. Perspectives and application of halophilic enzymes. In Halophiles. Sustainable Development and Biodiversity, vol 6 (eds. Maheshwari, D. & Saraf, M.) (Springer, 2015).
Leon, M. J. et al. Compatible solute synthesis and import by the moderate halophile Spiribacter salinus: Physiology and genomics. Front. Microbiol. 9, 108 (2018).
Santos, H. & da Costa, M. S. Compatible solutes of organisms that live in hot saline environments. Environ. Microbiol. 4(9), 501–509 (2002).
Gupta, S., Sharma, P., Dev, K. & Sourirajan, A. Halophilic bacteria of Lunsu produce an array of industrially important enzymes with salt tolerant activity. Biochem. Res. Int. 2016, 9237418. https://doi.org/10.1155/2016/9237418 (2016).
Mehta, V. J., Thumar, J. T. & Singh, S. P. Production of alkaline protease from an alkaliphilic actinomycetes. Bioresourc. Technol. 97, 1650–1654 (2006).
Vijayanand, S., Hemapriya, J., Selvinb, J. & Kiran, S. Production and optimization of haloalkaliphilic protease by an extremophile—Halobacterium sp. Js1, isolated from thalassohaline environment. Glob. J. Biotechnol. Biochem. 5(1), 44–49 (2010).
Reang, L. et al. Plant growth promoting characteristics of halophilic and halotolerant bacteria isolated from coastal regions of Saurashtra Gujarat. Sci. Rep. 12(1), 4699 (2022).
Shivanand, P., Mugeraya, G. & Kumar, A. Utilization of renewable agricultural residues for the production of extracellular halostable cellulase from newly isolated Halomonas sp. strain PS47. Ann. Microbiol. 63, 1257–1263 (2013).
Kumar, S., Karan, R., Kapoor, S., Singh, S. P. & Khare, S. K. Screening and isolation of halophilic bacteria producing industrially important enzymes. Braz. J. Microbiol. 43(4), 1595–1603 (2012).
Menendez, E., Garcia-Fraile, P. & Rivas, R. Biotechnological applications of bacterial cellulases. Bioengineering 2(3), 163–182 (2015).
Meticulous Market Research Pvt. Ltd. Industrial enzymes market worth $11.05 billion by 2029—exclusive report by Meticulous Research. https://www.globenewswire.com/en/news-release/2022/03/09/2400115/0/en/Industrial-Enzymes-Market-Worth-11-05-Billion-by-2029-Exclusive-Report-by-Meticulous-Research.html (2022).
Precedence Research. Industrial enzymes market (by product: Carbohydrases, proteases, lipases, polymerases & nucleases, others; by source: Plants, animals, microorganisms; by application: Food & beverages, detergents, animal feed, biofuels, nutraceutical, wastewater, others)—global industry analysis, size, share, growth, trends, regional outlook, and forecast 2021–2030. https://www.precedenceresearch.com/industrial-enzymes-market (2022).
Vijayaraghavana, P. & Prakash, V. G. S. Cow dung as a novel, inexpensive substrate for the production of a halo-tolerant alkaline protease by Halomonas sp. PV1 for eco-friendly applications. Biochem. Eng. J. 69, 57–60 (2012).
Vidyasagar, M., Prakash, S., Jayalakshmi, S. K. & Sreeramulu, K. Optimization of culture conditions for the production of halothermophilic protease from halophilic bacterium Chromohalobacter sp. TVSP101. World J. Microbiol. Biotechnol. 23, 655–662 (2007).
Lang, F. Mechanisms and significance of cell volume regulation. J. Am. Coll. Nutr. 26(5), 613S-623S. https://doi.org/10.1080/07315724.2007.10719667 (2007).
Roberts, M. F. Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Syst. 4, 1–5 (2005).
Detkova, E. N. & Boltyanskaya, Y. V. Osmoadaptation of haloalkaliphilic bacteria: Role of osmoregulators and their possible practical application. Microbiology 76(5), 511–522 (2007).
Reguera, M., Peleg, Z. & Blumwald, E. Targeting metabolic pathways for genetic engineering abiotic stress-tolerance in crops. Biochim. Biophys. Acta 1819(2), 186–194 (2012).
Jain, M. Emerging role of metabolic pathways in abiotic stress tolerance. J. Plant Biochem. Physiol. 1(2), 1–2 (2013).
Singh, M., Kumar, J., Singh, S., Singh, V. P. & Prasad, S. M. Roles of osmoprotectants in improving salinity and drought tolerance in plants: A review. Rev. Environ. Sci. Bio/Technol. 14(3), 407–426. https://doi.org/10.1007/s11157-015-9372-8 (2015).
Rajan, L. A. et al. Cloning and heterologous expression of ectoine biosynthesis genes from Bacillus halodurans in Escherichia coli. Biotechnol. Lett. 30(8), 1403–1407 (2008).
Anburajan, L., Meena, B. & Sivvaswamy, S. N. First report on molecular characterization of novel betaine aldehyde dehydrogenase from the halotolerant eubacteria, Bacillus halodurans. Gene Rep. 9, 131–135 (2017).
Kushner, D. J. & Kamekura, M. Physiology of halophilic eubacteria. In Halophilic Bacteria (ed. Rodriguez-Valera, F.) 109–140 (CRC Press, 1988).
Anithajothi, R., Nagarani, N., Umagowsalya, G., Duraikannu, K. & Ramakritinan, C. M. Screening, isolation and characterization of protease producing moderately halophilic microorganism Halomonas meridiana associated with coral mucus. Toxicol. Environ. Chem. 96(2), 296–306 (2014).
Sarker, P. K., Talukdar, S. A., Deb, P., Sayem, S. M. A. & Mohsina, K. Optimization and partial characterization of culture conditions for the production of alkaline protease from Bacillus licheniformis P003. SpringerPlus 2, 506 (2013).
Hadj-Ali, N. E. et al. Biochemical and molecular characterization of a detergent stable alkaline serine-protease from a newly isolated Bacillus licheniformis NH1. Enzyme Microb. Technol. 40, 515–523 (2007).
Potumarthi, R., Subhakar, C. & Jetty, A. Alkaline protease production by submerged fermentation in stirred tank reactor using Bacillus licheniformis NCIM-2042: Effect of aeration and agitation regimes. Biochem. Eng. J. 34, 185–192 (2007).
Nejad, Z. G., Yaghmaei, S. & Hosseini, R. H. Production of extracellular protease and determination of optimal condition by Bacillus licheniformis BBRC 100053. Int. J. Eng. 22(3), 221–228 (2009).
Benit, N., Biji, G. D. & Vijayaraghavan, P. Optimization of carboxymethyl cellulase production from Halomonas sp. isolated from saltpan. Int. J. Pharm. Biol. Sci. 8(3), 1025–1030 (2018).
da Silva, R. N., de Andrade Melo, L. F. & Finkler, C. L. L. Optimization of the cultivation conditions of Bacillus licheniformis BCLLNF-01 for cellulase production. Biotechnol. Rep. 29, e00599 (2021).
Gbenro, T., Adesanmi, A., Kamaraj, M., Saraswathi, K. & SindhuArumugam, F. P. Optimization of culture conditions for partially purified cellulase production by Oceanobacillus species isolated from wood industry soil in Chennai, India. Int. J. Sci. Eng. Res. 10(2), 384–389 (2019).
Akhir, S. M. et al. Media optimization of chitinase enzyme production from shrimp waste using Bacillus licheniformis TH-1 by response surface methods. Biotechnology 8(1), 120–125 (2009).
Keliat, J. M., Suryanto, D. & Munir, E. Characterization of extraceluller chitinase produced by Bacillus licheniformis JP2 from Penen Hot Springs, North Sumatera. Front. Eng. Manage. 2(5), 24–27 (2016).
Hussin, N. A. & Majid, A. H. A. Termiticidal activity of chitinase enzyme of Bacillus licheniformis, a symbiont isolated from the gut of Globitermes sulphureus worker. Biocatal. Agric. Biotechnol. 24(3), 101548 (2020).
Akeed, Y., Atrash, F. & Naffaa, W. Partial purification and characterization of chitinase produced by Bacillus licheniformis B307. Heliyon 6, e03858 (2020).
Sasi, A. et al. Identification and characterization of a newly isolated chitinase-producing strain Bacillus licheniformis SSCL-10 for chitin degradation. Hindawi Archaea 2020, 1–9 (2020).
Essghaier, B. et al. Characterization of a novel chitinase from a moderately halophilic bacterium, Virgibacillus marismortui strain M3–23. Ann. Microbiol. 62, 835–841 (2012).
Essghaier, B., Rouaissi, M., Boudabous, A., Jijakli, H. & Sadfi-Zouaoui, N. Production and partial characterization of chitinase from a halotolerant Planococcus rifitoensis strain M2–26. World J. Microbiol. Biotechnol. 26, 977–984 (2010).
Zhang, L. H., Lang, Y. J. & Nagata, S. Efficient production of ectoine using ectoine-excreting strain. Extremophiles 13(4), 717–724 (2009).
Van-Thuoc, D., Guzman, H., Quillaguaman, J. & Hatti-Kaul, R. High productivity of ectoines by Halomonas boliviensis using a combined two-step fed-batch culture and milking process. J. Biotechnol. 147(1), 46–51 (2010).
Van-Thuoc, D., Guzman, H., Thi-Hang, M. & Hatti-Kaul, R. Ectoine production by Halomonas boliviensis: Optimization using response surface methodology. Mar. Biotechnol. 12(5), 586–593 (2010).
Chen, Q., Zhang, L., Li, X., Liu, S. & Li, D. Poly-b-hydroxybutyrate/ectoine co-production by ectoine-excreting strain Halomonas salina. Process Biochem. 49, 33–37 (2014).
Chen, W. et al. Production and characterization of ectoine using a moderately halophilic strain Halomonas salina BCRC17875. J. Biosci. Bioeng. https://doi.org/10.1016/j.jbiosc.2017.12.011 (2017).
Meena, B., Anburajan, L., Ponni, B., Sivvaswamy, S. N. & Nandagopal, S. Highly potential compatible solute from halotolerant Bacillus subtilis MA04: Its functional and molecular characterization. Gene Rep. 4, 184–189 (2016).
Kunte, H. J., Truper, H. G. & Stan-Lotter, H. Halophilic microorganisms. In Astrobiology. The Quest for the Conditions of Life. Physics and Astronomy Online Library (eds. Horneck, G. & Baumstark-Khan, C.) 185–200 (Springer, 2002).
Canovas, D. et al. Isolation and characterization of salt-sensitive mutants of the moderate halophile Halomonas elongata and cloning of the ectoine synthesis genes. J. Biol. Chem. 272(41), 25794–25801 (1997).
Ono, H. et al. Accumulation of compatible solutes, ectoine and hydroxyectoine, in a moderate halophile, Halomonas elongata KS3 isolated from dry salty land in Thailand. J. Ferment. Bioeng. 85(4), 362–368 (1998).
Al-Gozyer, R. F. M., Moghaieb-Reda, E. A., Abdallah, A. A., Sharaf, A. N. & Abdallah, N. Characterization of salt tolerance in four halophytic bacteria strain isolated from solar saltern at Alexandria-Egypt. Biosci. Res. 15(3), 1905–1916 (2018).
Guzman, H., Van-Thuoc, D., Martin, J., Hatti-Kaul, R. & Quillaguaman, J. A process for the production of ectoine and poly-(3-hydroxybutyrate) by Halomonas boliviensis. Appl. Microbiol. Biotechnol. 84, 1069–1077 (2009).
Nakayama, H., Yoshida, K., Ono, H., Murooka, Y. & Shinmyo, A. Ectoine, the compatible solute of Halomonas elongata, confers hyperosmotic tolerance in cultured tobacco cells. Plant Physiol. 122, 1239–1247 (2000).
Moghaieb, R. E. A. et al. Characterization of salt tolerance in ectoine‐transformed tobacco plants (Nicotiana tabaccum): Photosynthesis, osmotic adjustment, and nitrogen partitioning. Plant Cell Env. 29, 173–182 (2006).
Moghaieb, R. E. A., Nakamura, A., Saneoka, H. & Fujita, K. Evaluation of salt tolerance in ectoine-transgenic tomato plants (Lycopersicum esculentum) in terms of photosynthesis, osmotic adjustment and carbon partitioning. GM Crops 2(1), 58–65 (2011).
Ishitani, M., Nakamura, T., Han, S. Y. & Takabe, T. Expression of the betaine aldehyde dehydrogenase gene in barley in response to osmotic stress and abscisic acid. Plant Mol. Biol. 27, 307–315 (1995).
Tian, F. et al. Overaccumulation of glycine betaine makes the function of the thylakoid membrane of wheat better under salt stress. Crop J. 5(1), 73–82 (2017).
Jackson, M. L. Soil Chemical Analysis (Prentice Hall of Indian Pvt. Ltd., 1973).
Walkley, A. & Black, I. A. An examination of methods for determining organic carbon and nitrogen in soils. J. Agric. Sci. 25, 589–609 (1935).
Olsen, S. R., Cole, C. V. Watanabe, F. S. & Dean, L. A. Estimation of available phosphorus in soil by extraction with sodium bicarbonate. In Circular of U.S. Department of Agriculture 939 (1954).
Cupp-Enyard, C. Sigma’s non-specific protease activity assay—Casein as a substrate. J. Visual. Exp. 19, 899 (2008).
Lay Mg Mg, Z., Than, W. M. & Myint, M. Study on the cellulase enzyme producing activity of bacteria isolated from manure waste and degrading soil. Int. J. Tech. Res. Appl. 3(6), 165–169 (2015).
Adney, B. & Baker, J. Measurement of cellulase activities. In Laboratory Analytical Procedure (LAP) (1996).
Hsu, S. C. & Lockwood, J. L. Powdered chitin agar as a selective medium for enumeration of actinomycetes in water and soil. Appl. Microbiol. 29, 422–426 (1975).
Setia, I. N. & Suharjono, N. Chitinolytic assay and identification of bacteria isolated from shrimp waste based on 16S rDNA sequences. Adv. Microbiol. 5, 541–548 (2015).
Acknowledgements
The author is sincerely thankful and highly indebted to the Department of Biotechnology and Food Testing Laboratory, Junagadh Agricultural University for their valuable support and research facilities provided and to all faculties, staff, and friends who were involved directly or indirectly in the successful accomplishment of the research.
Author information
Authors and Affiliations
Contributions
The first author Mr. L. Reang is the sole writer of the manuscript who conducted the research under the supervision of Dr. S. B. Bhatt (Main Corresponding Author). Dr. R. S. Tomar was an advisory committee member who guided in the molecular identification of the isolates. Dr. K. Joshi was an SRF and was involved in handling microbial culture-related works while Dr. S. Padhiyar, an SRF, assisted in the isolation, quantification, and purification of isolates’ gDNA and initial PCR screening works. Dr. H. Bhalani was a Research Associate and was involved in the PCR analysis of the studied genes while Dr. J.K. Kheni, a Research Associate assisted mainly in the bioinformatics analysis, and Dr. U. M. Vyas was the advisor of the sampling procedure while Dr. M. V. Parakhia was the lab in-charge involved in the arrangement of all the chemicals required. I, as the main corresponding author of this paper hereby state that the entire research work was carried out with the appropriate consent of all the concerned authors and that all the concerned authors in relation to this work described has given their approval for participation into this publication process.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Reang, L., Bhatt, S., Tomar, R.S. et al. Extremozymes and compatible solute production potential of halophilic and halotolerant bacteria isolated from crop rhizospheric soils of Southwest Saurashtra Gujarat. Sci Rep 14, 15704 (2024). https://doi.org/10.1038/s41598-024-63581-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63581-z
- Springer Nature Limited