
Abstract
Hypertrophic cardiomyopathy (HCM) is an inherited disorder characterized by left ventricular hypertrophy and diastolic dysfunction, and increases the risk of arrhythmias and heart failure. Some patients with HCM develop a dilated phase of hypertrophic cardiomyopathy (D-HCM) and have poor prognosis; however, its pathogenesis is unclear and few pathological models exist. This study established disease-specific human induced pluripotent stem cells (iPSCs) from a patient with D-HCM harboring a mutation in MYBPC3 (c.1377delC), a common causative gene of HCM, and investigated the associated pathophysiological mechanisms using disease-specific iPSC-derived cardiomyocytes (iPSC-CMs). We confirmed the expression of pluripotent markers and the ability to differentiate into three germ layers in D-HCM patient-derived iPSCs (D-HCM iPSCs). D-HCM iPSC-CMs exhibited disrupted myocardial sarcomere structures and an increased number of damaged mitochondria. Ca2+ imaging showed increased abnormal Ca2+ signaling and prolonged decay time in D-HCM iPSC-CMs. Cell metabolic analysis revealed increased basal respiration, maximal respiration, and spare-respiratory capacity in D-HCM iPSC-CMs. RNA sequencing also showed an increased expression of mitochondrial electron transport system-related genes. D-HCM iPSC-CMs showed abnormal Ca2+ handling and hypermetabolic state, similar to that previously reported for HCM patient-derived iPSC-CMs. Although further studies are required, this is expected to be a useful pathological model for D-HCM.
Similar content being viewed by others


Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited cardiac disorder characterized by ventricular wall hypertrophy and diastolic dysfunction. It is diagnosed by excluding secondary cardiomyopathies that result from various systemic diseases. In about 40–60% of cases, pathogenic mutations are found in genes encoding myocardial component proteins, primarily sarcomeres1,2. MYBPC3, which encodes cardiac myosin-binding protein C (cMYBP-C), is the most frequent causative gene for HCM along with MYH7, a β-myosin heavy chain-coding gene2. More than 350 MYBPC3 mutations that can cause HCM have been identified, and various MYBPC3 mutations have been identified in all regions3,4. The penetrance of HCM caused by MYBPC3 mutations increases with age, and the average penetrance in all generations is approximately 60%5. In HCM caused by MYH7 mutation, increased Ca2+ sensitivity, concomitant increased muscle contractility, and decreased muscle relaxation are present. Mutant sarcomere proteins often exhibit impaired Ca2+-dependent actomyosin cross-bridge cycling, altered Ca2+ sensitivity, and impaired force generation6. A similar mechanism has been estimated for HCM caused by MYBPC3 mutations, and haploinsufficiency has also been estimated as a mechanism of HCM pathogenesis because MYBPC3 mutations are mostly truncated7. However, the involvement of MYBPC3 mutations in the pathogenesis of HCM is not fully understood7,8,9.
Most patients with HCM have a relatively gradual disease course or may be asymptomatic; however, some progress to the dilated phase of hypertrophic cardiomyopathy (D-HCM), in which the left ventricular wall becomes thin and the ventricular chamber becomes large, resulting in systolic dysfunction10,11. D-HCM can cause heart failure, arrhythmias, and sudden death. Its symptoms, clinical course, and treatment are similar to that of dilated cardiomyopathy (DCM). However, D-HCM has a poorer prognosis than DCM and there is no fundamental treatment other than heart transplantation12,13. Moreover, there are few reports on the causes and pathological mechanisms of D-HCM in cellular and animal models.
In this study, we generated disease-specific human induced pluripotent stem cells (iPSCs) from a patient with MYBPC3 truncating mutation and analyzed the molecular cellular characteristics of disease-specific iPSC-derived cardiomyocytes (iPSC-CMs). This study aimed to explore the molecular mechanisms underlying the pathogenesis of D-HCM.
Results
Patient characteristics
The patient was diagnosed with impaired glucose tolerance approximately at age 50 years and was diagnosed with atrial fibrillation at 53 years. The patient’s father was diagnosed with and treated for heart failure and atrial fibrillation; however, other detailed information was unknown. At 57 years, edema and shortness of breath appeared when using the stairs. Echocardiography showed asymmetric interventricular septal hypertrophy with normal ventricular dimensions and an ejection fraction (EF) (EF 66%) (Fig. 1A). Based on the results of the cardiac catheterization, the patient was diagnosed with HCM. At the age of 60 years, he developed ventricular fibrillation and underwent cardioverter-defibrillator implantation. Subsequently, a decrease in left ventricular systolic function and ventricular dilatation were observed (EF 34%) (Fig. 1A). Genetic analysis using panel sequencing revealed a frameshift mutation (c.1377delC, p.L460Wfs*5) in MYBPC3, but no other pathogenic variants. Histological staining of the endomyocardial biopsy samples revealed myocardial degeneration and severe fibrosis (Fig. 1B). Based on clinical findings and genetic analysis, the patient was diagnosed with D-HCM. The patient underwent standard medical therapy for heart failure, and multiple catheter ablation procedures for atrial fibrillation and ventricular tachycardia. However, the patient’s health condition progressively worsened and he died of heart failure at the age of 65 years.

Echocardiograms and pathological images in a patient with D-HCM. (A). Diastolic (left) and systolic (right) echocardiograms at 57 (above) and 64 years old (below), respectively. LVDd: left ventricular end-diastolic diameter, LVDs: left ventricular end-systolic diameter, EF: ejection fraction, IVST: interventricular septal thickness, PWT: posterior LV wall thickness. (B). Histological staining of endomyocardial biopsy sample: Hematoxylin–eosin stain (HE: left) and Masson trichrome stain (MT: right) (Scale: 200 µm (Full), 50 µm (Enlarged)).
Generation of D-HCM iPSCs
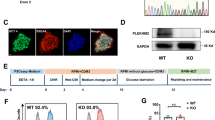
The established D-HCM iPSCs formed colonies and proliferated as shown in Fig. 2A. Direct DNA sequencing of D-HCM iPSCs revealed the same mutation (c.1377delC) in MYBPC3 as that in the donor blood sample (Fig. 2B). iPSCs were highly stained for the pluripotency markers Nanog, Oct3/4, and Sox2 (Fig. 2C). Flow cytometry using the stem cell markers SSEA-4 and TRA-1-60 showed that 88.0% of D-HCM iPSCs were positive for both markers (Fig. 2D). To confirm whether D-HCM iPSCs had differentiation potential in all three germinal directions, we performed immunofluorescence using the germ markers TUJ1 (ectoderm), SMA (mesoderm), and AFP (endoderm). The D-HCM iPSCs stained positive for all three germ markers. (Fig. 2E). From these results, we confirmed the generation of disease-specific iPSCs from the PBMCs of a D-HCM patient with the MYBPC3 mutation (c.1377delC).

Pluripotency of the D-HCM iPSCs and cardiac differentiation of iPSCs into iPSC-CMs. (A). D-HCM iPSCs form several colonies (scale,100 µm). (B). DNA sequencing analysis of D-HCM iPSCs reveals MYBPC3 mutation c.1377delC (red arrow). (C). Immunofluorescence with three pluripotency markers (Nanog, Oct3/4, and Sox2) (scale:50 µm). (D). Flow cytometry of two pluripotency markers (SSEA-4 and TRA-1–60). (E). Immunofluorescence with three germ layer markers: ectoderm (TUJ1), mesoderm (αSMA), endoderm (AFP) (Scale:100 µm). (F). Cardiac induction was performed using the GiWi protocol (Day0-14), purification with a lactate medium (Day15-20), and maturation with a fatty acid medium (Day22-30). In the lower part, photographs show each stage of cardiac differentiation on Day 0, Day 5, Day 10, and Day 50 (Scale:200 µm).
Cardiac characteristics in iPSC-CMs
Autonomous beating was observed approximately one week after cardiac induction (Fig. 2F). Cardiac sarcomere structures of iPSC-CMs were observed using fluorescent immunostaining for cardiac troponin T (cTnT) (Fig. 3A). Flow cytometry revealed that more than 80% of the control- (Ctrl) and D-HCM iPSC-CMs were positive for cTnT (Fig. 3B). The maturity of iPSC-CMs was assessed by fluorescent immunostaining for MLC2-A (immature and atrial muscle markers) and MLC2-V (ventricular muscle marker). Generally, MLC2-V is selectively expressed in ventricular CMs, whereas MLC2-A is expressed in both atrial and ventricular CMs. Moreover, the expression of MLC2-A in immature ventricular CMs gradually decreased with CMs maturation14. Although some iPSC-CMs co-expressed MLC2-A and MLC2-V, more than 50% of iPSC-CMs were MLC2-V dominant (Ctrl:55.0%; D-HCM:55.9%) (Fig. 3C,D). c-MYBP-C protein and MYBPC3 mRNA expression levels in iPSC-CMs were not significantly different between the control and D-HCM groups (Fig. 3E,F). Moreover, troponin I (TnI) protein expression was also not significantly different between the control and D-HCM groups (Supplementary Figure S1).

Cardiac characteristics of iPSC-CMs. (A). Immunofluorescence for Cardiac Troponin T (cTnT) (Scale: 50 µm). (B). Flow cytometry with cTnT (blue wave: negative control, red wave: cTnT positive). (C). Immunofluorescence for MLC2-A and MLC2-V (Scale: 50 µm). blue: DAPI, green: MLC2-A, red: MLC2-V. (D). Fluorescence-positive cell count (%) for MLC2-A and MLC2-V in Fig. 2C. iPSC-CMs were classified into three groups according to the expression levels, V > A, V≒A, V < A (V: MLC2-V, A: MLC2-A). (E). Protein expression level of cMYBP-C (Ctrl: N = 4 from 2 cell lines, D-HCM: N = 5 from 1 cell line). (F). mRNA expression level of MYBPC3 (Ctrl: N = 6 from 2 cell lines, D-HCM: N = 6 from 1 cell line).
Increased myocardial sarcomere disarray and mitochondrial damage in D-HCM iPSC-CMs
The intracellular myocardial structures of iPSC-CMs were evaluated using TEM, and cardiac sarcomere structures were clearly observed in both the Ctrl- and D-HCM groups (Fig. 4A). Most sarcomeres were regularly arranged in the Ctrl group, whereas some sarcomeres in the D-HCM group showed Z-band disruption and sarcomere disarray (Fig. 4A, white arrows). Sarcomere length did not differ between the Ctrl group (1.65 ± 0.02 μm) and the D-HCM group (1.64 ± 0.02 μm) (Fig. 4B). The Z-band disruption rate, measured in sarcomere units, was 1.4% in the Ctrl group and 14.7% in the D-HCM group (Fig. 4C), and this difference was statistically significant. Next, we evaluated the mitochondrial structure in iPSC-CMs. Cristae are internal mitochondrial structures and its loss indicates mitochondrial damage15. As shown in Fig. 4D, cristae loss area per mitochondrion was significantly higher in D-HCM (13.6 ± 1.1%) than in the Ctrl group (4.2 ± 0.37%) (Fig. 4E). Additionally, the mitochondrial area was larger in the D-HCM (0.35 ± 0.04 μm2) than in the Ctrl group (0.24 ± 0.03 μm2) (Fig. 4F).

Electron microscopy images of cardiac sarcomeres and mitochondria in iPSC-CMs. (A). Cardiac sarcomere structures. Z-band disruption and sarcomere disarray were observed in D-HCM iPSC-CMs (white arrow) (Scale: 5 µm (upper), 2 µm (middle), 1 µm (lower)). (B). Sarcomere lengths (µm) (Ctrl: N = 71, D-HCM: N = 72). (C). Z-band disruption rate (%) measured by sarcomere units (Ctrl: N = 140, D-HCM: N = 220). “N” represents the sarcomere units in A, B, and C. (D). Mitochondrial structures. More damaged mitochondria were observed in D-HCM iPSC-CMs (white arrow) (Scale: 500 nm). (E). The cristae loss area (%) indicates damaged mitochondria (Ctrl: N = 72, D-HCM: N = 68). (F). Mitochondrial size (um2) (Ctrl: N = 53, D-HCM: N = 42). “N” represents the numbers of mitochondria in D, E, and F. All results of TEM were obtained from Ctrl 1 cell line and D-HCM 1 cell line. n.s. indicates not significant vs. Ctrl; **p < 0.01 vs. Ctrl; ***p < 0.001 vs. Ctrl.
Increased abnormal Ca2+ transients in D-HCM iPSC-CMs
The representative Ca2+ signals and transients are shown in Fig. 5A. Compared to Ctrl iPSC-CMs, abnormal Ca2+ signals (early after depolarization (EAD) and delayed after depolarization (DAD)) increased in the D-HCM iPSC-CMs both before and after 1 µM isoproterenol (iso) treatment. The percentages of cells with abnormal Ca2+ transients were 4.8% in the Ctrl, 24.0% in the D-HCM, 5.0% in the Ctrl treated with iso, and 15.4% in the D-HCM treated with iso (Fig. 5B). The peak time was prolonged in D-HCM iPSC-CMs after iso treatment (Fig. 5C), and the decay time was significantly prolonged in D-HCM iPSC-CMs with or without iso treatment (Fig. 5D).

Intracellular Ca2+imaging in iPSC-CMs. (A). Ca2+ transients before and after isoproterenol (iso) treatment. Abnormal Ca2+ transients were increased in D-HCM iPSC-CMs (red arrow). (B). Number of cells with abnormal Ca2+ transients (%). N = 21 (Ctrl), N = 25 (D-HCM), N = 20 (Ctrl iso), N = 26 (D-HCM iso). “N” represents the number of cells. (C). Time to peak (second: sec) (D). 50% decay time (sec). n.s. indicates not significant vs. Ctrl; **p < 0.01 vs. Ctrl; ***p < 0.001 vs. Ctrl.
Increased mitochondrial energy production and metabolic shift beginning in D-HCM iPSC-CMs
The mitochondrial stress test was used to assess mitochondrial aerobic respiration (Fig. 6A), and a Real-Time ATP Rate Assay was used to assess the ratio of aerobic to anaerobic respiration in ATP production. Basal respiration, maximal respiration, and spare respiratory capacity significantly increased in D-HCM iPSC-CMs, suggesting accelerated mitochondrial energy production (Fig. 6B,C,D). ATP production tended to be higher in D-HCM iPSC-CMs; however, this difference was not statistically significant (Fig. 6E). Real-time ATP rate assays showed that mitochondrial ATP production was predominant in Ctrl iPSC-CMs, whereas glycolytic ATP production was predominant in D-HCM iPSC-CMs and was significantly higher than that in the Ctrl group (Fig. 6F). RT-qPCR showed that the expression of the glucose transporter GLUT4 was upregulated in D-HCM iPSC-CMs (Fig. 6G). These results suggested increased mitochondrial energy production and the beginning of the metabolic shift to glycolysis in D-HCM iPSC-CMs.

Mitochondrial and glycolytic energy production in iPSC-CMs. (A). Cell Mito Stress Test (blue: Ctrl, red: D-HCM). Black lines indicate the addition of mitochondrial functional modulators (Oligomycin, FCCP, Rotenone/Antimycin A). Vertical axis: oxygen consumption rate (OCR), horizontal axis: time. (B–E). Energy production indicators in mitochondrial respiration calculated from Fig. 5A. (B). Basal respiration. (C). Maximal respiration. (D). Spare respiratory capacity. (F). Mitochondrial and glycolytic ATP production rate measured by Real-Time ATP Rate Assay. The left graph shows the ATP production rate (pmol/min/50 K Cells). The right graph shows the ATP production rate (%). glyco ATP: glycolytic ATP production (anaerobic respiration), mito ATP: mitochondrial ATP production (aerobic respiration). (G). mRNA expression levels of GLUT4 by RT-qPCR. n.s. indicates not significant vs. Ctrl; *p < 0.05 vs. Ctrl; ***p < 0.001 vs. Ctrl.
Upregulated mitochondria-related gene expressions in D-HCM iPSC-CMs
RNA sequencing was performed to examine the differences in gene expression patterns between Ctrl and D-HCM iPSC-CMs. According to enrichment analysis using the GSEA software16, mitochondria-related gene expression was enriched in D-HCM iPSC-CMs. In GO analysis, the top-ranked gene sets in D-HCM iPSC-CMs were closely related to ATP production and mitochondrial metabolism (Fig. 7A,B). The heatmap in Fig. 7C shows that gene expression of the electron transport system Complex I-V in the mitochondria was elevated in D-HCM iPSC-CMs. Among the mitochondrial electron transport system-related genes, the expression levels of NDUFB1, COX5B and ATPIF1 were significantly increased in D-HCM iPSC-CMs, as confirmed by RT-qPCR (Fig. 7D).

Gene expression pattern in D-HCM iPSC-CMs. (A). GSEA analysis using Gene Ontology (GO). Top-ranked gene set lists were arranged in each table (p < 0.05 and low FDR rank). Ctrl: N = 4 from 2 cell lines, D-HCM: N = 5 from 1 cell line. (B). Graphs of the top two gene sets in GO. (C). Heatmap of mitochondrial electron transport-related genes. (D). mRNA expression levels of mitochondrial electron transport-related genes: NDUFB1, COX5B, UQCRB and ATPIF1 by RT-qPCR. n.s. indicates not significant vs. Ctrl; *p < 0.05 vs. Ctrl; **p < 0.01 vs. Ctrl.
Discussion
In this study, we established iPSCs from PBMCs of a D-HCM patient carrying MYBPC3 truncating mutation (c.1377delC), compared the phenotypes of Ctrl iPSC-CMs and D-HCM iPSC-CMs generated by cardiac differentiation and obtained the following findings: (1) TEM showed that D-HCM iPSC-CMs had a more disorganized sarcomere structure and intramitochondrial cristae defects. (2) Ca2+ imaging showed that D-HCM iPSC-CMs exhibited more abnormal Ca2+ signals and longer decay times. (3) Cell metabolic analysis showed that mitochondrial energy production increased, and energy metabolism began to switch to glycolytic dominance in D-HCM iPSC-CMs. Furthermore, (4) RNA-seq and RT-qPCR showed that the expression levels of oxidative phosphorylation- and ATP production-related genes were upregulated in D-HCM iPSC-CMs. This is the first study to show the pathophysiological phenotypes of disease-specific iPSC-CMs established from a patient with D-HCM and may be a useful tool for elucidating the pathogenesis of D-HCM and develo** new therapeutic strategies.
c-MYBP-C binds to myosin and actin, regulates their positions, and plays a role in maintaining normal cardiac sarcomere structure and regulating normal contractions3,8,17. All C0-10 domains of cMYBP-C are required for normal cardiac function, and various pathogenic mutations in each domain have been reported as the causative genes of HCM. The MYBPC3 c.1377delC mutation identified in this study resulted in a frameshift and truncated protein, p.L460Wfs*5 (located in the C3 domain17), which has previously been reported as a causative gene mutation in HCM. This variant has not been identified in the general population and is presumed to result in an absent or nonfunctional protein product, although functional assays have not been conducted. Although D-HCM-specific pathogenic variants are unknown, previous studies have reported that some MYBPC3 pathogenic variants, such as p.R945fs and p.R820Q, are detected in patients with D-HCM18,19. Pathogenic mutations in MYBPC3 are presumably responsible for impaired myosin-actin binding and abnormal cardiac sarcomere structures resulting in excessive contraction20,21,22. Disarrangement of the sarcomere structure has also been observed in patients with HCM carrying mutations in other sarcomere genes such as MYH7, which encodes the cardiac myosin heavy chain, and may be a common finding in HCM with sarcomere-related gene mutations23,24,25. Most MYBPC3 mutations that cause HCM are of the short form, and haploinsufficiency has been suggested as a mechanism of pathogenesis7. In this study, MYBPC3 mRNA and protein levels were not significantly different between Ctrl iPSC-CMs and D-HCM iPSC-CMs. Previous studies have reported that MYBPC3 mRNA and protein expression is decreased or unchanged in iPSC-CMs with MYBPC3 mutations, depending on the type and site of MYBPC3 mutations7,26,27.
In this study, the size of D-HCM iPSC-CMs was not larger than that of Ctrl iPSC-CMs. A previous study reported that the cell size was indeed larger in HCM iPSC-CMs with MYBPC3 mutations than in Ctrl iPSC-CMs; however, in the presence of serum, cardiomyocyte hypertrophy was masked and rather smaller in HCM iPSC-CMs with MYBPC3 mutation28. Another study also reported that HCM iPSC-CMs with MYBPC3 mutations showed no significant difference in cell size compared to Ctrl iPSC-CMs27. This may be because iPSC-CMs mimic cardiomyocytes at the developmental stage and thus reflect the early prehypertrophic stage of HCM29.
In this study, Ca2+ imaging showed abnormal Ca2+ handling in D-HCM iPSC-CMs. The prolonged decay time of D-HCM iPSC-CMs may reflect diastolic dysfunction24,25,30, which is a characteristic clinical finding in HCM. In addition, the increased abnormal Ca2+ transients in D-HCM iPSC-CMs suggested triggered activity, including EAD and/or DAD, which are important mechanisms in the pathogenesis of arrhythmias. Intracellular Ca2+-handling abnormalities are common pathophysiological features of HCM observed in disease-specific iPSC-CMs, animal models, and human tissue samples, including markedly increased intracellular Ca2+ levels, prolonged Ca2+ transient, and triggered activity31. HCM-causing sarcomeric mutations are thought to increase myofilament Ca2+ sensitivity, myocardial contractility, and impair myocardial energetics, resulting in a hypermetabolic mitochondrial state and abnormal Ca2+ handling32.
In a previous study, increased basal respiration and ATP production were observed in genome-edited MYH7 mutant HCM iPSC-CMs, which is consistent with our results33. In contrast, another report showed that ATP production decreased in myocardial samples from patients with HCM who underwent septal myectomy15. Although this discrepancy may be attributed to differences in the disease stage or the analyzed samples, the relative increase in energy demand seems to be a common feature of HCM34. The increased mitochondrial respiratory capacity of D-HCM iPSC-CMs observed in our cellular energy metabolism analysis was presumably due to a compensatory mechanism for abnormal sarcomeric functional changes and excess energy demand, which was predicted to increase oxidative stress and cause mitochondrial damage. This suggests that D-HCM iPSC-CMs exhibit a phenotype similar to HCM in terms of cellular energy metabolism22. In this study, cristae loss was frequently observed in D-HCM iPSC-CMs by TEM. Excessive mitochondrial energy production is predicted to increase oxidative stress and cause mitochondrial damage15. In addition, since most mitochondria in cardiomyocytes are arranged near sarcomeres and provide energy for myocardial contraction, it is possible that the alteration in sarcomere structures directly affects mitochondrial function via the mitochondrial network35. Furthermore, some reports have shown that the failure to upregulate mitophagy is partly responsible for the accumulation of damaged mitochondria in HCM36,37.
In this study, D-HCM iPSC-CMs showed elevated expression of glycolysis-related genes GLUT4 and ATPIF1 and glycolysis-dominant energy metabolism, suggesting metabolic remodeling of the D-HCM state. A recent study demonstrated that ATPIF1 upregulation contributes to the switch from oxidative metabolism to glycolysis in the heart during the development of pathological hypertrophy, and that deletion of ATPIF1 in cardiomyocytes or scavenging of mitochondrial ROS prevents the metabolic switch and protects against pathological remodeling38. Since the transition from oxidative fatty acid metabolism to glucose metabolism is thought to be associated with pathological remodeling of the heart30, the metabolic switch and accumulation of mitochondrial damage may contribute to the transition to the dilated phase in patients with HCM.
This study had some limitations. We used only one patient-derived iPSC-CM and compared it with iPSC-CMs from two unrelated healthy subjects as controls. In the future, it would be desirable to establish more iPSCs from patients with D-HCM and create isogenic controls for comparison. Although iPSC-CMs are useful and non-invasive tools for exploring pathological mechanisms and discovering novel therapeutic targets for heart diseases, cardiomyocyte immaturity is a limitation in the application of iPSC-CMs for research purposes39,40. In this study, we used the fatty acid medium method to accelerate iPSC-CM maturation41. The phenotype of D-HCM iPSC-CMs was similar to that of the pathological models of HCM reported in previous studies22,23,25,26,33. Therefore, we were unable to completely elucidate the precise molecular mechanisms of transition to D-HCM. Further studies are needed to elucidate the detailed pathophysiological mechanisms underlying D-HCM.
In conclusion, disease-specific iPSCs (D-HCM iPSCs) were established from a patient with D-HCM and a shortened MYBPC3 mutation (c.1377delC). D-HCM iPSC-CMs showed an abnormal cardiac sarcomere structure, increased damaged mitochondria, abnormal Ca2+ transients, increased mitochondrial energy production, and upregulated glycolysis-related gene expression. D-HCM iPSC-CMs showed a phenotype similar to that of a previously reported pathological model of HCM; however, further studies are needed to elucidate the pathological mechanism of D-HCM.
Methods
All experiments were performed in accordance with the latest version of the Declaration of Helsinki and the relevant guidelines and regulations including the Ethical Guidelines for Medical and Health Research Involving Human Subjects. The generation and experiments of iPSC were reviewed and approved by the University of Tsukuba Clinical Research Ethics Review Committee (approval number: R02-078). The study on the genetic analysis was also reviewed and approved by the University of Tsukuba Clinical Research Ethics Review Committee (approval number: R02-300).
Generation of iPSC from a patient with D-HCM
iPSCs were generated as described in our previous paper42. Briefly, after obtaining written informed consent from the patient, the peripheral blood mononuclear cells (PBMCs) were collected from the patient’s peripheral blood through density gradient centrifugation using Ficoll-Plaque and were cultured with PBMC medium (Supplemental Table). After one week, Human iPS Cell Generation Episomal Vector Mix (TaKaRa) was transfected into the PBMCs by electroporation using the Neon Transfection System (Thermo Fisher Scientific), and cells were quickly spread onto the iMatrix-coated plates with StemFit medium (Supplemental Table) supplemented 10 µM Y-27632 (FUJIFILM Wako). About two weeks later, iPSC colonies were picked under a microscope, and D-HCM patient-derived iPSCs (D-HCM iPSCs) were established. Two cell lines, HPS3354 and HPS3386, were purchased from the RIKEN BioResource Center and used as healthy control cells (Ctrl iPSCs).
Genetic analysis
After obtaining written informed consent from the patient, next-generation sequencing was performed with the Ion Proton System (Thermo Scientific, Waltham, Massachusetts, USA) using the Ion AmpliSeq™ Cardiovascular Research Panel and the Ion AmpliSeq™ Library Kit 2.0. Primary processing of reads was performed using the Ion Proton Software (Thermo Fisher Scientific). Alignment with the reference genome (GRCh38–hg19), coverage analysis, and variant calling were performed using standard parameters in the Ion Torrent Software Suite (ISS) version 5.4.0. The VCF file was uploaded and annotated using wANNOVAR software. Variants classified as pathogenic or likely pathogenic for DCM, HCM, and familial atrial fibrillation according to the ClinVar were considered as causative genes, which were confirmed by direct sequencing.
Cardiac differentiation
Cardiac differentiation was performed according to the GiWi (GSK-3β (glycogen synthase kinase-3β) inhibitor and Wnt inhibitor) protocol41,43, and mediums and reagents are listed in the Supplemental Table. iPSCs were cultured with StemFit in a 5% CO2 incubator at 37℃. When the cells reached 80–90% confluence, the medium was replaced with RPMI (Supplemental Table) containing 8 µM CHIR99021 (a GSK3β inhibitor, Sigma-Aldrich). Twenty-four hours later (Day 1), the medium was replaced with RPMI, and the cells were cultured. Forty-eight hours later (Day 3), the medium was changed with RPMI- medium + 2 µM Wnt-C59 (an Wnt inhibitor, Cayman Chemical Company). On Day 5, the medium was replaced with RPMI- medium. On day 7, the medium was replaced with the RPMI + medium (Supplemental Table). After these processes, the medium was replaced with RPMI + every two days. During days 12–14, the cells were passaged on Matrigel-coated 6-well plates with 10% FBS medium (Supplemental Table) supplemented with 10 µM Y-27362. Two days later, the medium was replaced with lactate (Supplemental Table) to remove non-iPSC-CMs. Five days later, the medium was replaced with 10% FBS medium. Two or three days later, the medium was replaced with fatty acid medium (Supplemental Table) for cell maturation. Approximately one week later, the medium was replaced with 10% FBS medium, and the medium was replaced every three days. iPSC-CMs were used in our experiments approximately 50 days after cardiac differentiation (Days 45–55).
Immunofluorescence
Immunofluorescence analysis was performed as previously described42. Briefly, cells on a Matrigel-coated cover glass were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich). After washing, cells were blocked with 1% BSA. The primary antibody was reacted overnight at 4 °C. The next day, the cells on the cover glass were incubated with a secondary antibody for one hour at room temperature. After washing, nuclear staining was performed, and the samples were sealed. The images were acquired using a confocal laser microscope TCS SP8 (Leica Microsystems). The primary antibodies used were a Human Pluripotent Stem Cell 3-Color Immunocytochemistry Kit (R&D Systems), Anti-Cardiac Troponin T antibody (1:200, Richard-Allan Scientific), Anti-MLC-2 V antibody (1:200, Proteintech), and anti-MLC-A antibody (1:200, Synaptic Systems). Secondary antibodies used were Alexa Fluor 488 goat anti-mouse IgG (1:2000) and Alexa Fluor 555 goat anti-rabbit IgG (1:2000; Thermo Fisher Scientific). DAPI (300 nM) and FluoroPure (Thermo Fisher Scientific) were used for nuclear staining.
Flow cytometry
Flow cytometry was performed as described previously42. Briefly, the collected cells were stained using the Live/Dead Fixable Dead Cell Stain Kit (1:1000; Thermo Fisher Scientific). The samples were fixed and permeabilized using Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent (Thermo Fisher Scientific), respectively. The primary antibody was diluted in permeabilization buffer and incubated for 30 min at room temperature. After washing, the cells were incubated with secondary antibody for 20 min at room temperature. The samples were suspended in an auto-MACS buffer and passed through a filter. The samples were analyzed using the BD FACSVerse System (BD Biosciences) and FlowJo (BD Biosciences). The primary antibodies used were PE anti-human SSEA-4 (1:100; BioLegend), TRA-1-60 anti-human Vio 488 (1:100; Miltenyi Biotec), and Anti-Cardiac Troponin T (1:200; Richard-Allan Scientific). Secondary antibodies included Alexa Fluor 488 goat anti-mouse IgG (1:2000; Thermo Fisher Scientific).
Differentiation in three germ layers
An in vitro differentiation assay was performed as described previously42. Briefly, D-HCM iPSCs suspension was seeded into 96U Bottom Plate (Thermo Fisher Scientific) and cultured with StemFit supplemented 10 µM Y-27632. To form an embryoid body (EB), the medium was changed to DMEM (Thermo Fisher Scientific) supplemented with 4 g/L D( +)-Glucose Solution (FUJIFILM Wako), 10% FBS, and 1% P/S (penicillin–streptomycin solution (FUJIFILM Wako)) every two days. After eight days, the EBs were transferred onto 0.1% gelatin-coated 12-well plates. After culturing for one week, immunostaining was performed and the differentiation potential into three germ layers was evaluated using a fluorescence microscope BZ-X710 (KEYENCE). The antibodies used were anti-mouse TUJ1 (1:100; R&D Systems), anti-mouse SMA (1:100; R&D Systems), anti-mouse AFP (1:100; R&D Systems), and Alexa Fluor 488 goat anti-mouse IgG (1:2000; Thermo Fisher Scientific).
Transmission electron microscope: TEM
iPSC-CMs were seeded onto Matrigel-coated plates covered with a cover glass. Prefixation was performed with 2.5% glutaraldehyde/0.1 M phosphate buffer for 30 min at room temperature. Samples were post-fixed, embedded, and polymerized at the Medical Electron Microscopy Laboratory of the Department of Medicine, University of Tsukuba. The ultrathin sections were placed on copper grids, imaged using a transmission electron microscope JEM-1400 (JEOL), and analyzed using ImageJ44,45.
Ca2+ imaging
Ca2+ imaging was performed as described in our previous paper46. Briefly, iPSC-CMs were seeded onto Matrigel-coated glass-bottom dishes. The day before measurement, the medium was replaced with Ca2+ imaging medium (Supplemental Table). Just before the measurements, cells were incubated with 5 µM Fluo 4 AM special packaging (Do**do) for 10 min in a 37 °C incubator. The intracellular Ca2+ kinetics of iPSC-CMs were measured using line scans on a confocal laser microscope (ZEISS) and analyzed using ImageJ software. To measure the time to peak and 50% decay time, we chose records of Ca2+ transients in which no EAD/DAD appeared, and then measured the time from baseline to peak and the time from peak to 50% descent. After baseline recording, the samples were treated with 1 µM isoproterenol (iso) (β-receptor agonist, Kowa) and measured in the same way.
Western blot
Western blotting was performed as described previously42. Briefly, proteins were extracted from the collected cells using the PRO-PREP Protein Extraction Solution (INB). Proteins were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to PVDF membranes, and incubated in blocking buffer (0.1% Tris-buffered saline (Santa Cruz) supplemented with 3% skim milk (FUJIFILM Wako) and 0.1% polysorbate 20 (MP Biomedicals)) for one hour at room temperature. Next, the primary antibody diluted with blocking buffer was reacted overnight at 4 °C. The following day, the secondary antibody was diluted in the blocking buffer and reacted for one hour at room temperature. After washing, the target proteins were detected using the ELC Prime Western Blotting Detection Reagent (Cytiva) and photographed using the chemiluminescence imaging system FUSION FX7.EDGE (Vilber Bio Imaging). The ImageJ software was used for statistical analysis. The antibodies used were against MYBPC3 antibody (E-7) (1:1000; Santa Cruz Biotechnology), Troponin I antibody (4002S) (1:1000; Cell Signaling) and actin antibody (C4) (1:1000; Santa Cruz Biotechnology). The cMYBP-C protein levels were calculated by standardization with β-actin.
Cell metabolic analyses
The Cell Mito Stress Test and Real-Time ATP Rate Assay were performed using a Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies), according to the manufacturer’s instructions. Four to five days before the measurement, iPSC-CMs were seeded onto Matrigel-coated XFp plates. On the day of the measurement, the medium was replaced with Seahorse XF RPMI Medium. The measurement reagents were 1.5 µM Oligomycin (oxidative phosphorylation inhibitor), 1 µM FCCP (deconjugating agent), and 0.5 µM Rotenone/Antimycin A (electron transfer system inhibitors). Seahorse Waves software (Agilent Technologies) were used for all the analyses.
Reverse transcription quantitative polymerase chain reaction: RT-qPCR
RT-qPCR was performed as previously described46. Briefly, total RNA was purified from cell pellets using the RNeasy Mini Kit (QIAGEN). A High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) was used to synthesize cDNA from each RNA sample. qPCR was performed using Quant Studio 5 (Thermo Fisher Scientific) and the standard curve method. GAPDH primer (Integrated DNA Technologies) was used as an internal control. MYBPC3, GLUT4, NDUFB1, COX5B, UQCRB, and ATPIF1 (Integrated DNA Technologies) were used as target primers.
RNA sequencing
RNA was purified from cell pellets using a RNeasy Mini Kit (QIAGEN). After the samples were prepared, RNA sequencing was performed at the Department of Sports Medicine of the Organization for Open Facility Initiatives of the University of Tsukuba47. NGS was performed using a NextSeq 500 System (Illumina), and NGS data were confirmed using CLC Genomics Workbench 22.0 software (QIAGEN). Gene Set Enrichment Analysis (GSEA) software was used for RNA sequencing analysis, using the Gene Ontology (GO) gene set16. A heatmap was drawn using Morpheus software (https://software.broadinstitute.org/morpheus/).
Statistical analyses
Values for each group are presented as mean ± standard error. Two-tailed t-tests or Mann–Whitney U tests were used to test 2-group comparisons. p < 0.05 was considered statistically significant (ns > 0.05, * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001). All analyses were performed using GraphPad Prism software.
Data availability
All data generated and/or analyzed in the current study are available from the corresponding author upon reasonable request. The total RNA-Seq data as FASTQ files and an expression browser as table data have been deposited in the “Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra; accessed on 3 Apr 2024)” under accession number: SRP499665.
References
Maron, B. J. et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 79, 372–389. https://doi.org/10.1016/j.jacc.2021.12.002 (2022).
Marian, A. J. & Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 121, 749–770 (2017).
Carrier, L., Mearini, G., Stathopoulou, K. & Cuello, F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573, 188–197. https://doi.org/10.1016/j.gene.2015.09.008 (2015).
Marston, S., Copeland, O., Gehmlich, K., Schlossarek, S. & Carrrier, L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy?. J. Muscle Res. Cell Motility. 33, 75–80. https://doi.org/10.1007/s10974-011-9268-3 (2012).
Page, S. P. et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: Disease expression in relation to age, gender, and long term outcome. Circ. Cardiovasc. Genet. 5, 156–166 (2012).
Marian, A. J. Molecular genetic basis of hypertrophic cardiomyopathy. Circ. Res. 128, 1533–1553 (2021).
Van Dijk, S. J. et al. Cardiac myosin-binding protein C mutations and hypertrophic ardiomyopathy haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483 (2009).
Harris, S. P., Lyons, R. G. & Bezold, K. L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 108, 751–764 (2011).
Li, J., Feng, X. & Wei, X. Modeling hypertrophic cardiomyopathy with human cardiomyocytes derived from induced pluripotent stem cells. Stem. Cell Res. Therapy. https://doi.org/10.1186/s13287-022-02905-0 (2022).
Aizawa, Y. et al. Incidence, clinical characteristics, and long-term outcome of the dilated phase of hypertrophic cardiomyopathy. Keio J. Med. 68, 87–94 (2019).
Killu, A. M. et al. Cardiac resynchronization therapy in patients with end-stage hypertrophic cardiomyopathy. Europace 20, 82–88 (2018).
Hamada, T. et al. Clinical features of the dilated phase of hypertrophic cardiomyopathy in comparison with those of dilated cardiomyopathy. Clin. Cardiol. 33, E24 (2010).
Goto, D. et al. Clinical characteristics and outcomes of dilated phase of hypertrophic cardiomyopathy: Report from the registry data in Japan. J. Cardiol. 61, 65–70 (2013).
Fukushima, H. et al. Specific induction and long-term maintenance of high purity ventricular cardiomyocytes from human induced pluripotent stem cells. PLoS One 15, e0241287 (2020).
Ranjbarvaziri, S. et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 144, 1714–1731 (2021).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. https://doi.org/10.1073/pnas.0506580102 (2005).
Sadayappan, S. & de Tombe, P. P. Cardiac myosin binding protein-C: Redefining its structure and function. Biophys. Rev. 4, 93–106. https://doi.org/10.1007/s12551-012-0067-x (2012).
Konno, T. et al. A novel missense mutation in the myosin binding protein-C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J. Am. Coll. Cardiol. 41, 781–786 (2003).
Hitomi, N. et al. A frameshift deletion mutation in the cardiac myosin-binding protein C gene associated with dilated phase of hypertrophic cardiomyopathy and dilated cardiomyopathy. J. Cardiol. 56, 189–196 (2010).
McNamara, J. W. et al. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS One 12, e0180064 (2017).
Toepfer, C. N. et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 11, eaat1199 (2019).
Toepfer, C. N. et al. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation https://doi.org/10.1161/CIRCULATIONAHA.119.042339 (2020).
Tanaka, A. et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 3, e001263 (2014).
Zhou, W. et al. Induced pluripotent stem cell-derived cardiomyocytes from a patient with MYL2-R58Q-mediated apical hypertrophic cardiomyopathy show hypertrophy, myofibrillar disarray, and calcium perturbations. J. Cardiovasc. Transl. Res. 12, 394–403 (2019).
Han, L. et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 104, 258–269 (2014).
Qiu, H. et al. Inhibition of HSC70 alleviates hypertrophic cardiomyopathy pathology in human induced pluripotent stem cell-derived cardiomyocytes with a MYBPC3 mutation. Clin. Transl. Med. 11, e647 (2021).
Helms, A. S. et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight 5, e133782 (2020).
Dambrot, C. et al. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell. Mol. Med. 18, 1509–1518 (2014).
Pioner, J. M. et al. Slower calcium handling balances faster cross-bridge cycling in human MYBPC3 HCM. Circ. Res. 132, 628–644 (2023).
Viola, H. M. & Hool, L. C. Impaired calcium handling and mitochondrial metabolic dysfunction as early markers of hypertrophic cardiomyopathy. Arch. Biochem. Biophys. 665, 166–174. https://doi.org/10.1016/j.abb.2019.03.006 (2019).
Coppini, R., Santini, L., Olivotto, I., Ackerman, M. J. & Cerbai, E. Abnormalities in sodium current and calcium homoeostasis as drivers of arrhythmogenesis in hypertrophic cardiomyopathy. Cardiovasc. Res. 116, 1585–1599. https://doi.org/10.1093/cvr/cvaa124 (2020).
Ferrantini, C. et al. Mechanical and energetic consequences of HCM-causing mutations. J. Cardiovasc. Transl. Res. 2, 441–451 (2009).
Mosqueira, D. et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 39, 3879–3892 (2018).
Aung, L. H. H., Jumbo, J. C. C., Wang, Y. & Li, P. Therapeutic potential and recent advances on targeting mitochondrial dynamics in cardiac hypertrophy: A concise review. Mol. Therapy Nucl. Acids 25, 416–443. https://doi.org/10.1016/j.omtn.2021.06.006 (2021).
Katti, P. et al. Mitochondrial network configuration influences sarcomere and myosin filament structure in striated muscles. Nat. Commun. 13, 6058 (2022).
Ong, S. B. & Hausenloy, D. J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 88, 16–29. https://doi.org/10.1093/cvr/cvq237 (2010).
Van Der Velden, J. et al. Metabolic changes in hypertrophic cardiomyopathies: Scientific update from the working group of myocardial function of the European Society of Cardiology. Cardiovasc. Res. 114, 1273–1280. https://doi.org/10.1093/cvr/cvy147 (2018).
Zhou, B. et al. Upregulation of mitochondrial ATPase inhibitory factor 1 (ATPIF1) mediates increased glycolysis in mouse hearts. J. Clin. Invest. https://doi.org/10.1172/JCI155333 (2022).
Andrysiak, K., Stępniewski, J. & Dulak, J. Human-induced pluripotent stem cell-derived cardiomyocytes, 3D cardiac structures, and heart-on-a-chip as tools for drug research. Pflugers Archiv. Eur. J. Physiol. 473, 1061–1085. https://doi.org/10.1007/s00424-021-02536-z (2021).
Knight, W. E. et al. Maturation of pluripotent stem cell-derived cardiomyocytes enables modeling of human hypertrophic cardiomyopathy. Stem. Cell Rep. 16, 519–533 (2021).
Horikoshi, Y. et al. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells 8, 1095 (2019).
Shimoda, Y. et al. Generation of a human induced pluripotent stem cell line derived from a patient with dilated cardiomyopathy carrying LMNA nonsense mutation. Stem. Cell Res. 62, 102793 (2022).
Lian, X. et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175 (2013).
Lam, J. et al. A universal approach to analyzing transmission electron microscopy with imagej. Cells 10, 2177 (2021).
Gherghiceanu, M. et al. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell Mol. Med. 15, 2539–2551 (2011).
Aonuma, K. et al. Novel preventive effect of isorhamnetin on electrical and structural remodeling in atrial fibrillation. Clin. Sci. 136, 1831–1849 (2022).
Sugasawa, T., Kanki, Y., Komine, R., Watanabe, K. & Takekoshi, K. Identification of RNA markers in red blood cells for do** control in autologous blood transfusion. Genes (Basel) 13, 1255 (2022).
Acknowledgements
This study was supported in part by grants from the Tsukuba Basic Research Support Program to N.M.[FY2020, Type B]. There are no financial relationships related to the work presented in this manuscript. We would like to thank Mrs. Yumi Isaka and Mrs. Sayaka Inoue for their assistance with the experiments. We would also like to thank Editage (www.editage.com) for the English language editing.
Author information
Authors and Affiliations
Contributions
H.M. and N.M. designed the study and prepared the manuscript. H.M., D.X., Y.S., and N.M. generated iPSCs and performed all the experiments. D.X. supervised Ca2+ imaging, and Z.Y. supervised the RNA sequencing analysis. Y.M., B.X., and K.T. provided experimental and analytical support. K.S., M.Y., and T.I. collected clinical information. M.I. and N.M. reviewed the experiments and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mori, H., Xu, D., Shimoda, Y. et al. Metabolic remodeling and calcium handling abnormality in induced pluripotent stem cell-derived cardiomyocytes in dilated phase of hypertrophic cardiomyopathy with MYBPC3 frameshift mutation. Sci Rep 14, 15422 (2024). https://doi.org/10.1038/s41598-024-62530-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62530-0
- Springer Nature Limited