
Abstract
Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) has recently been attributed to biallelic repeat expansions in RFC1. More recently, the disease entity has expanded to atypical phenotypes, including chronic neuropathy without cerebellar ataxia or vestibular areflexia. Very recently, RFC1 expansions were found in patients with Sjögren syndrome who had neuropathy that did not respond to immunotherapy. In this study RFC1 was examined in 240 patients with acute or chronic neuropathies, including 105 with Guillain-Barré syndrome or Miller Fisher syndrome, 76 with chronic inflammatory demyelinating polyneuropathy, and 59 with other types of chronic neuropathy. Biallelic RFC1 mutations were found in three patients with immune-mediated neuropathies, including Guillain-Barré syndrome, idiopathic sensory ataxic neuropathy, or anti-myelin-associated glycoprotein (MAG) neuropathy, who responded to immunotherapies. In addition, a patient with chronic sensory autonomic neuropathy had biallelic mutations, and subclinical changes in Schwann cells on nerve biopsy. In summary, we found CANVAS-related RFC1 mutations in patients with treatable immune-mediated neuropathy or demyelinating neuropathy.
Similar content being viewed by others

Introduction
Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) has been recently found to be caused by biallelic repeat expansions in the intron of RFC11. More recently, the disease entity has expanded to chronic neuropathy without cerebellar ataxia or vestibular areflexia2. Repeat configuration includes AAGGG, ACAGG, AGGGC, or combinations thereof without clear genotype and phenotype relationship2,3,4.
Immune-mediated neuropathy can be categorized into acute and chronic. Guillain-Barré syndrome (GBS) or Miller Fisher syndrome (MFS) is representative of acute immune-mediated neuropathy, whereas chronic inflammatory demyelinating polyneuropathy (CIDP) is representative of chronic immune-mediated neuropathy. Myelin-associated glycoprotein (MAG) neuropathy, originally derived from CIDP, is characterized by sensory and autonomic neuropathy with positive serum monoclonal IgM antibody against MAG5 and is at least partly treatable by immunotherapy including rituximab6. Notably, neuropathies cannot be solely attributed to the presence of antibodies. In fact, our previous study showed that among two siblings who simultaneously suffered from Campylobacter jejuni-associated diarrhea and subsequently tested positive for serum anti-ganglioside antibodies, only one developed GBS, suggesting the involvement of some unknown host factors7.
Our previous study showed that CAG repeat expansions in ATXN2, which causes spinocerebellar ataxia type 2 (SCA2), were found in patients with CIDP or immune-mediated neuropathy8. A recent review also suggested the association between repeat expansions and various diseases, including immune-mediated diseases9. Very recently, RFC1 expansions were found in patients with Sjögren syndrome who had neuropathy that did not respond to immunotherapy, suggesting that neuropathy could be attributed to RFC1 expansions10. Thus, a previous study suggested that unnecessary immunotherapy should be avoided in patients who have immune-mediated diseases with RFC1 mutations10. However, we speculate that immunotherapy might be effective in some patients with certain unknown conditions. We therefore hypothesized that repeat expansion may be a background genetic factor for immune-mediated neuropathy. Accordingly, the current study examined RFC1 in 240 patients with immune-mediated neuropathy, such as GBS, CIDP, and MAG neuropathy, as well as other types of neuropathies.
Patients and methods
Patients
This study enrolled Japanese patients and controls from the Kinki region of Japan between 2005 and 2023. RFC1 was analyzed in 240 Japanese patients with neuropathy (146 men and 94 women; mean age, 54 ± 18 years), including 105 with GBS or MFS (62 men and 43 women, mean age, 50 ± 19 years), 76 patients with CIDP (44 men and 32 women, mean age, 56 ± 17 years), and 59 with other types of chronic neuropathy (40 men and 19 women, mean age, 61 ± 17 years), which included eight patients with MAG neuropathy, four with paraneoplastic syndrome (without MAG antibody), three with Sjögren syndrome, and three with multifocal neuropathy. Patients with known genetic causes were excluded. The clinical diagnosis was established after the first admission for diagnostic workup when DNA samples were collected. Chronic neuropathy was herein defined as weakness or sensory disturbance in two or more limbs over at least 2 months with other signs or symptoms being minimal and corresponding abnormalities in nerve conduction studies (NCS). A control group consisting of 160 apparently healthy participants (90 men and 70 women; mean age, 64 ± 17 years) was also included. The nerve biopsy specimen was analyzed as described previously11.
Genetic analyses
DNA was extracted from peripheral blood using the Qiagen Kit (Qiagen, Hilden Germany). Primers used for the amplification of the short range of the repeat region in RFC1 were as described. When no normal size band was detected, the sample was subjected to repeat-primed polymerase chain reaction (PCR) for AAGGG (pathogenic), ACAGG (pathogenic), AGGGC (pathogenic), AGAGG (possibly pathogenic), AAGGC (possibly pathogenic), AAAGG (variable penetrance), AAAAG (likely non-pathogenic), AAAGGG (likely non-pathogenic), and AAGAG (likely non-pathogenic) repeat configurations1,2,4. Primers used for repeat-primed PCR are described in supplemental material. All patients provided written informed consent for genetic analyses. All methods in this study were performed in accordance with the Declaration of Helsinki. The study protocol was approved by the Institutional Review Boards of Kagoshima University and Kindai University.
Autoantibody measurement
Anti-ganglioside antibodies were examined in patients with GBS, MFS, CIDP, and IgM paraproteinemic neuropathy using GM1, GM2, GM3, GD1a, GD1b, GD3, GT1b, and GQ1b as reported previously12,13. Anti-GalNAc-GD1a antibody was additionally examined in patients with GBS and MFS, and anti-GT1a antibody was examined when anti-GQb1 antibody was detected12,13. Anti-MAG antibody and glycolipid sulfoglucuronyl paragloboside (SGPG) antibody, cross-reactive to MAG, were examined in patients with IgM paraproteinemic neuropathy as reported previously5. All methods in this study were performed in accordance with the Declaration of Helsinki. The study protocol was approved by the Institutional Review Boards of Kindai University.
Results
RFC1 analyses
RFC1 mutations were found in three patients with immune-mediated neuropathy, such as GBS, MAG neuropathy, and idiopathic sensory ataxic neuropathy and one patient with sensory autonomic neuropathy (Fig. 1). No expansion of AGGGC, AGAGG, AAGGC, AAAGG, AAAAG, AAAGGG, or AAGAG was found in the four patients. No mutations were found in the control group.

Repeat-primed PCR results for RFC1. Pathological repeats of AAGGG or ACAGG were expanded in patients with Guillain-Barré syndrome, idiopathic sensory ataxic neuropathy with mild motor deficit (ISAN), MAG neuropathy, or sensory autonomic neuropathy with mild motor deficit (SAN).
Clinical information of patients with RFC1 mutations
Clinical and genetic information of patients with RFC1 mutations is summarized in Table 1. Detailed clinical information is provided upon request. Results of NCS are summarized in Supplementary Table 1.
Nerve biopsy findings of patient 4
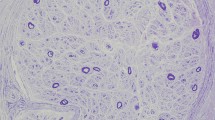
Sural nerve biopsy revealed loss of myelinated and unmyelinated nerves (Fig. 2A and B), a typical finding for CANVAS-related neuropathy1. Amyloidosis was excluded by Congo red staining (not shown). An electron microscopic image revealed many collagen pockets (Fig. 2C). Notably, electron microscopic analysis of Schwann cells revealed cytoplasmic inclusion bodies, dense material, or accumulated membranous material (Fig. 2D–G). No apparent abnormality was found in the vascular systems (Fig. 2H).

Sural nerve biopsy findings in Patient 4. (A) A light microscopic image showing marked loss of myelinated fibers, without amyloidosis or vasculitis. (B) A lager image showing marked loss of both large and small myelinated fibers. (C) An electron microscopic image showing many collagen pockets (black arrow), reflecting unmyelinated fiber damage. (D) A Schwann cell had cytoplasmic dense materials (blue arrow). (E) Another Schwann cell had a cytoplasmic inclusion (yellow arrow). (F) The other Schwann cell had a cytoplasmic inclusion similar to the one shown in E (yellow arrow). (G) Membranous materials were seen between myelin sheaths (white arrow) or between a myelin sheath and an axon (white arrowhead). (H) Cells that constitute vessels had no abnormality without aggregation.
Discussion
We found biallelic RFC1 mutations in three patients with immune-mediated neuropathy and one with non-immune mediated neuropathy. This has been the first study to show an association between repeat expansions in RFC1 and treatable immune-mediated neuropathy. Patient 1 had typical monophasic GBS with anti-ganglioside antibodies wherein weakness was completely resolved after IVIG, but dysesthesia in the upper limbs remained 3 years after disease onset. Patient 2, who suffered from idiopathic sensory ataxic neuropathy with mild motor deficit, had a recurrent and remission clinical course with some improvement in motor and sensory conditions after repeated IVIG treatment. Patient 3 had MAG demyelinating neuropathy with stable symptoms and decreased IgM levels after rituximab treatment. All three patients tested positive for autoantibodies. Although positivity for rheumatoid factor, IgM anti-IgG, found in patient 2 might not directly cause neuropathy, a previous study demonstrated that rheumatic patients with rheumatoid factor had significantly more frequent neuropathies than those without rheumatoid factor (83% vs. 44%)14. Common symptoms in the patients included sensory disturbances, similar to those in Patient 4 with chronic sensory autonomic neuropathy with mild motor deficit and in previous studies on CANVAS2.
Pathological studies for CANVAS have been limited, with electron microscopic analyses being reported in only one study15. While the reported electron microscopic findings were limited to descriptions regarding Schwann cells associated with unmyelinated axons, we found several rare findings concerning myelinating Schwann cells, with a cytoplasmic inclusion body, dense materials, or accumulated membranous materials. Although Patient 4 did not have electrophysiological evidence of demyelinating neuropathy, subclinical changes in Schwann cells may have occurred. In Patient 2, demyelination was suggested to have occurred in the sural nerve on biopsy at the early stage and in the tibial nerves on NCS at the late stage. In addition, Patient 3 with MAG neuropathy had demyelination in the median nerve on NCS. These findings may indicate that patients with RFC1 mutations occasionally develop demyelination neuropathy or Schwann cell damage.
A possible mechanism by which RFC1 mutations are associated with immune-mediated neuropathy includes the vulnerability of the nerves themselves or that of Schwann cells to autoantibodies. In fact, we showed subclinical abnormal findings in Schwann cells producing myelin as mentioned above. Another possible mechanism is the abnormal function of blood–nerve barrier, given that the blood–nerve barrier may protect the nervous system from toxic materials, including autoantibodies16. However, we found no abnormalities around blood vessels, the site of the blood–nerve barrier, though the absence of morphological changes cannot rule out its functional change. Alternatively, abnormal immune-response or production of autoantibodies may be promoted in such patients. For example, neuronal or myelin antigens, or an expanded repeat RNA, might be abnormally exposed as immunogens17. Despite these fascinating mechanisms, the association between RFC1 mutations and immune-mediated neuropathy remains inconclusive, because the relatively low incidence of mutations among a large cohort of studied patients may raise the possibility that the association is coincidental.
Biallelic mutations may suggest loss of function in RFC1, which is involved in DNA repair18. A recent report describing expansion mutations in an allele and truncation mutations in another supports this mechanism19. Loss of DNA repair protein function in neuropathy and cerebellar ataxia is reminiscent of Aprataxin-related disorders20. However, a previous study did not find any evidence suggesting loss of DNA repair function of RFC1 in fibroblasts1. Our electron microscopic findings suggested cytoplasmic inclusions in Schwann cells, a situation similar to that of SCA2-related neuropathy, a gain of function disease8. Further accumulation of evidence is needed to clarify the pathomechanism using neuronal or glial cell models or animal models.
One limitation of this study is the small number of patients with immune-mediated neuropathy who were positive for RFC1 mutations. In addition, detailed pathological studies in patients with immune-mediated neuropathy were lacking, as mentioned above.
In summary, we found CANVAS-related RFC1 mutations in patients with treatable immune-mediated neuropathy or demyelinating neuropathy. Thus, immunotherapy should not be terminated solely based on the identification of RFC1 mutations; however, repeated immunotherapy to unresponsive patients should be avoided10,21. Nonetheless, future large-scale studies are needed before definitive conclusions can be established.
Data availability
Detailed clinical information of patients with RFC1 mutations were available in the supplemental material.
References
Cortese, A. et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658 (2019).
Ando, M. et al. Genetic and clinical features of cerebellar ataxia with RFC1 biallelic repeat expansions in Japan. Front. Neurol. 13, 952493 (2022).
Yuan, J. H. et al. Multi-type RFC1 repeat expansions as the most common cause of hereditary sensory and autonomic neuropathy. Front. Neurol. 13, 986504 (2022).
Dominik, N. et al. Normal and pathogenic variation of RFC1 repeat expansions: Implications for clinical diagnosis. Brain https://doi.org/10.1093/brain/awad240 (2023).
Hamada, Y. et al. Binding specificity of anti-HNK-1 IgM M-protein in anti-MAG neuropathy: Possible clinical relevance. Neurosci. Res. 91, 63–68 (2015).
Parisi, M. et al. Efficacy of rituximab in anti-myelin-associated glycoprotein demyelinating polyneuropathy: Clinical, hematological and neurophysiological correlations during 2 years of follow-up. Eur. J. Neurol. 29, 3611–3622 (2022).
Hirano, M. et al. A family with Campylobacter enteritis: Anti-GD1a antibody with/without Guillain-Barre syndrome. Neurology 60, 1719–1720 (2003).
Inada, R. et al. Phenotypic and molecular diversities of spinocerebellar ataxia type 2 in Japan. J. Neurol. 268, 2933–2942 (2021).
Fotsing, S. F. et al. The impact of short tandem repeat variation on gene expression. Nat. Genet. 51, 1652–1659 (2019).
Fernandez-Eulate, G. et al. Sjogren syndrome and RFC1-CANVAS sensory ganglionopathy: Co-occurrence or misdiagnosis?. J. Neurol. 270, 460–465 (2023).
Oka, N., Kawasaki, T., Unuma, T., Shigematsu, K. & Sugiyama, H. Different profiles of onion bulb in CIDP and CMT1A in relation to extracellular matrix. Clin. Neuropathol. 32, 406–412 (2013).
Kusunoki, S. et al. N-acetylgalactosaminyl GD1a is a target molecule for serum antibody in Guillain-Barre syndrome. Ann. Neurol. 35, 570–576 (1994).
Yamagishi, Y. et al. Markers for Guillain-Barre syndrome with poor prognosis: A multi-center study. J. Peripher. Nerv. Syst. 22, 433–439 (2017).
Biswas, M. et al. Prevalence, types, clinical associations, and determinants of peripheral neuropathy in rheumatoid patients. Ann. Indian Acad. Neurol. 14, 194–197 (2011).
Magy, L. et al. Early diagnosis in cerebellar ataxia, neuropathy, vestibular Areflexia syndrome (CANVAS) by focusing on major clinical clues: Beyond ataxia and vestibular impairment. Biomedicines 10, 2046 (2022).
Kanda, T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 84, 208–212 (2013).
Wang, E. T. et al. What repeat expansion disorders can teach us about the central Dogma. Mol. Cell 83, 324–329 (2023).
Dilley, R. L. et al. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58 (2016).
Ronco, R. et al. Truncating variants in RFC1 in cerebellar ataxia, neuropathy, and vestibular Areflexia syndrome. Neurology 100, e543–e554 (2023).
Hirano, M. et al. DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann. Neurol. 61, 162–174 (2007).
Curro, R. et al. RFC1 expansions are a common cause of idiopathic sensory neuropathy. Brain 144, 1542–1550 (2021).
Acknowledgements
This study was partly supported by KAKEN (22K07510 to MH), 2023 Kindai University Research Enchancement Grant (KD2306 to MH), KAKEN (20K07894 to MK), KAKEN (21K15702 and 23K06931 to MA), Transformative Research Areas (A) (Multifaceted Proteins) (20H05927 to YN) from Ministry of Education, Culture, Sports, Science and Technology (MEXT), Scientific Research (B) (21H02840 to YN) from Japan Society for the Promotion of Science (JSPS), Practical Research Projects for Rare/Intractable Diseases (JP21ek0109532 to YN) from Japan Agency for Medical Research and Development (AMED), and Research on Measures for Intractable Diseases (20FC1041 to YN) from the Ministry of Health, Labour and Welfare (MHLW).
Author information
Authors and Affiliations
Contributions
M.H., M.K., KS., H.T., S.K. and Y.N. contributed to the conception and design of the study; Y.Y., M.S., K.F., S.Y., M.A., N.O., M.N., T.M., T.T. and K.S. contributed to the acquisition and analysis of data; M.H. and N.Y. contributed to drafting the text or preparing the figures.
Corresponding author
Ethics declarations
Competing interests
MH, MS, HT, and YN have received honoraria from Takeda. MH has received a research grant unrelated to this study from Takeda. HT and SK received honoraria from Japan Blood Product Organization. HT and SK have received honoraria from CSL Behring. All other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hirano, M., Kuwahara, M., Yamagishi, Y. et al. CANVAS-related RFC1 mutations in patients with immune-mediated neuropathy. Sci Rep 13, 17801 (2023). https://doi.org/10.1038/s41598-023-45011-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45011-8
- Springer Nature Limited