
Abstract
To understand the epidemiological and genetic characteristics of B19V, a multiple-province surveillance of patients with febrile rash illnesses (FRIs) were conducted in China during 2009 ~ 2021. The clinical specimens of 3,820 FRI patients were collected and tested for B19V DNA. A total of 99 (2.59%) patients were positive for B19V, and 49 (49.49%) were children under 5 years old. B19V infections occurred throughout the year without obvious seasonal pattern. Ten NS1-VP1u sequences and seven genome sequences were obtained in this study, identified as subgenotype 1a. Combined with the globally representative genome sequences, no temporal and geographic clustering trends of B19V were observed, and there was no significant correlation between B19V sequences and clinical manifestations. The evolutionary rate of the B19V genome was 2.30 × 10–4 substitutions/site/year. The number of negative selection sites was higher than that of positive selection sites. It was the first to comprehensively describe the prevalence patterns and evolutionary characteristics of B19V in FRI patients in China. B19V played the role in FRI patients. Children under 5 years old were the main population of B19V infection. Subgenotype 1a was prevalent in FRI patients in China. B19V showed a high mutation rate, while negative selection acted on the genome.
Similar content being viewed by others


Introduction
As a member of the family Parvoviridae, human parvovirus B19 (B19V) was a global and common infectious pathogen in humans1. B19V was first identified by Cossart et al.2 during the evaluation of tests for hepatitis B virus surface antigen in 1975. Subsequently, B19V infection has been reported worldwide. The transmission of B19V infection usually occurs through the respiratory route, and infections can also be transmitted vertically from mother to fetus and through the transfusion of blood products and bone marrow transplants3,4,5. B19V infection is common in childhood, and it can also occur throughout adulthood albeit at a lower rate6. The findings showed a significant negative correlation between viremia rates and age, and the positive rate of B19V DNA decreased with age, from 2.24% in 19–30 years to 0.87% in 41–50 years7,8. However, the prevalence of IgG antibodies directed against B19V ranges from 2 to 15% in children 1 to 5 years old, 15 to 60% in children 6 to 19 years old, 30 to 60% in adults, and more than 85% in the geriatric population9,10,11,12. The manifestations of B19V infection depend on the age, immunity and hematologic status of the host1,13. Most B19V infections are asymptomatic. The common clinical manifestation of B19V infection is erythema infectiosum in children, while arthropathy is a more common manifestation of infection in adults, particularly in women14. In immunocompromised hosts, persistent B19V infection presents with pure red cell aplasia and chronic anemia13,15. B19V is now recognized as the only etiologic agent of erythema infectiosum16,17,18. Due to similar eruption symptoms, erythema infectiosum can be confused with rubella19,20,21,22.
B19V is a small nonenveloped single-stranded DNA virus with an approximately 5.5-kb long genome. This DNA encodes six viral proteins, among which the three major proteins are nonstructural protein 1 (NS1), viral protein 1 (VP1) and viral protein 2 (VP2). In addition, there are three small nonstructural proteins of 7.5 kDa, X, and 11 kDa. Based on the phylogenetic analysis of the NS1-VP1u region, B19V was classified into three genotypes: genotype 1, genotype 2, and genotype 323. With the increasing number of genome sequences obtained, B19V was further subdivided into subgenotypes, which were useful for molecular epidemiological studies. Genotype 1 was further divided into two subgenotypes, 1a and 1b, and genotype 3 was divided into two subgenotypes, 3a and 3b24,25.
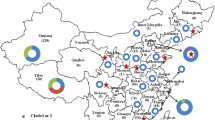
To date, investigations of B19V have been carried out by many researchers in recent years, including in China. However, most of them focused on viral infection in blood products from healthy donors, organ transplantation and the status of B19V infection in pregnant women26,27,28,29,30. There have been few studies on the prevalence of B19V in patients with febrile rash illnesses (FRIs). From January 2009 to June 2021, active surveillance of FRIs was conducted in ten provinces of China, including Anhui, Bei**g, Hebei, Henan, Hunan, Shandong, Shanxi, Shaanxi, Shanghai and **-stone sampling (SS). The best fit models for the genome and six proteins of B19V were determined by Bayesian factor analysis. The Markov Chain Monte Carlo method was performed for 50 million generations and sampled so that 1,000 trees were generated. Finally, the convergence of the chains and the effective sample size (> 200) were determined by Tracer software (version 1.7.1), and the uncertainty of the parameter estimates was assessed by a 95% HPD interval.
Selection pressure was also analyzed in six proteins of B19V based on the genome dataset. DnaSP6 software (Version 6.0) was used to calculate the ω values (ω = dN/dS) of B19V, where dN represents the nonsynonymous substitution rate and dS represents the synonymous substitution rate. Gene-specific and site-specific selection pressures were measured as the value dN/dS at each codon site and estimated using four different codon-based maximum-likelihood methods (FEL, SLAC, FUBAR and MEME), and Tamura-Nei model (TrN) or Hasegawa-Kishino-Yano model (HKY85) were used as nucleotide substitution models50. All methods were obtained available at the DataMonkey online version of the Hyphy package (www.datamonkey.org) with significance levels set at p < 0.05, and the posterior probability of the FUBAR algorithm was > 0.95.
Accession number
Seven genome sequences and three NS1-VP1u sequences of B19V in this study were submitted to GenBank with accession numbers OR533486 ~ OR533495 (Supplementary Table S1).
Ethics approval and consent to participate
This study was approved by the ethical review committee of the National Institute for Viral Disease Control and Prevention, Chinese Centers for Disease Control and Prevention. The informed consents were signed by patients or their legal guardians.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
Heegaard, E. D. & Brown, K. E. Human parvovirus B19. Clin. Microbiol. Rev. 15, 485–505. https://doi.org/10.1128/CMR.15.3.485-505.2002 (2002).
Cossart, Y. E., Field, A. M., Cant, B. & Widdows, D. Parvovirus-like particles in human sera. Lancet 1, 72–73. https://doi.org/10.1016/s0140-6736(75)91074-0 (1975).
Azzi, A., Morfini, M. & Mannucci, P. M. The transfusion-associated transmission of parvovirus B19. Transfus. Med. Rev. 13, 194–204. https://doi.org/10.1016/s0887-7963(99)80033-9 (1999).
Broliden, K. Parvovirus B19 infection in pediatric solid-organ and bone marrow transplantation. Pediatr. Transplant 5, 320–330. https://doi.org/10.1034/j.1399-3046.2001.00035.x (2001).
Rogo, L. D., Mokhtari-Azad, T., Kabir, M. H. & Rezaei, F. Human parvovirus B19: a review. Acta Virol. 58, 199–213. https://doi.org/10.4149/av_2014_03_199 (2014).
Young, N. S. & Brown, K. E. Parvovirus B19. N Engl. J. Med. 350, 586–597. https://doi.org/10.1056/NEJMra030840 (2004).
Abdelrahman, D., Al-Sadeq, D. W., Smatti, M. K., Taleb, S. A., AbuOdeh, R. O., Al-Absi E. S., et al. Prevalence and Phylogenetic Analysis of Parvovirus (B19V) among Blood Donors with Different Nationalities Residing in Qatar. Viruses, 13: (2021) doi:https://doi.org/10.3390/v13040540.
Kooistra, K., Mesman, H. J., de Waal, M., Koppelman, M. H. G. M. & Zaaijer, H. L. Epidemiology of high-level parvovirus B19 viraemia among Dutch blood donors, 2003–2009. Vox Sang. 100, 261–266. https://doi.org/10.1111/j.1423-0410.2010.01423.x (2011).
Anderson, L. J. et al. Detection of antibodies and antigens of human parvovirus B19 by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 24, 522–526. https://doi.org/10.1128/jcm.24.4.522-526.1986 (1986).
Cohen, B. J. & Buckley, M. M. The prevalence of antibody to human parvovirus B19 in England and Wales. J. Med. Microbiol. 25, 151–153. https://doi.org/10.1099/00222615-25-2-151 (1988).
Kelly, H. A. et al. The age-specific prevalence of human parvovirus immunity in Victoria Australia compared with other parts of the world. Epidemiol Infect 124, 449–457. https://doi.org/10.1017/s0950268899003817 (2000).
Tsujimura, M. et al. Human parvovirus B19 infection in blood donors. Vox. Sang. 69, 206–212. https://doi.org/10.1111/j.1423-0410.1995.tb02595.x (1995).
Qiu, J., Soderlund-Venermo, M. & Young, N. S. Human Parvoviruses. Clin. Microbiol. Rev. 30, 43–113. https://doi.org/10.1128/CMR.00040-16 (2017).
White, D. G. et al. Human parvovirus arthropathy. Lancet 1, 419–421. https://doi.org/10.1016/s0140-6736(85)91145-6 (1985).
El-Mahallawy, H. A., Mansour, T., El-Din, S. E., Hafez, M. & Abd-el-Latif, S. Parvovirus B19 infection as a cause of anemia in pediatric acute lymphoblastic leukemia patients during maintenance chemotherapy. J. Pediatr. Hematol. Oncol. 26, 403–406. https://doi.org/10.1097/00043426-200407000-00001 (2004).
Anderson, M. J., Lewis, E., Kidd, I. M., Hall, S. M. & Cohen, B. J. An outbreak of erythema infectiosum associated with human parvovirus infection. J. Hyg. (Lond) 93, 85–93. https://doi.org/10.1017/s0022172400060964 (1984).
Naides, S. J. Erythema infectiosum (fifth disease) occurrence in Iowa. Am. J. Public Health 78, 1230–1231. https://doi.org/10.2105/ajph.78.9.1230 (1988).
Plummer, F. A. et al. An erythema infectiosum-like illness caused by human parvovirus infection. N Engl. J. Med. 313, 74–79. https://doi.org/10.1056/NEJM198507113130203 (1985).
Yermalovich, M. A., Hubschen, J. M., Semeiko, G. V., Samoilovich, E. O. & Muller, C. P. Human parvovirus B19 surveillance in patients with rash and fever from Belarus. J. Med. Virol. 84, 973–978. https://doi.org/10.1002/jmv.23294 (2012).
Rezaei, F. et al. Prevalence and genotypic characterization of Human Parvovirus B19 in children with measles- and rubella-like illness in Iran. J. Med. Virol. 88, 947–953. https://doi.org/10.1002/jmv.24425 (2016).
Los de Angeles Ribas, M.,Tejero, Y.,Cordero, Y.,Perez, D.,Sausy, A.,Muller, C. P., et al. Identification of human parvovirus B19 among measles and rubella suspected patients from Cuba. J. Med. Virol., 91:1351-1354. (2019) doi:https://doi.org/10.1002/jmv.25444.
Grolhier, C. et al. When a viral eruption hides another one: intrafamilial outbreak of parvovirus B19 and measles virus co-infections: case report. BMC Infect Dis. 20, 496. https://doi.org/10.1186/s12879-020-05183-4 (2020).
Servant, A. et al. Genetic diversity within human erythroviruses: identification of three genotypes. J. Virol. 76, 9124–9134. https://doi.org/10.1128/jvi.76.18.9124-9134.2002 (2002).
Toan, N. L. et al. Phylogenetic analysis of human parvovirus B19, indicating two subgroups of genotype 1 in Vietnamese patients. J. Gen. Virol. 87, 2941–2949. https://doi.org/10.1099/vir.0.82037-0 (2006).
Parsyan, A., Szmaragd, C., Allain, J. P. & Candotti, D. Identification and genetic diversity of two human parvovirus B19 genotype 3 subtypes. J. Gen. Virol. 88, 428–431. https://doi.org/10.1099/vir.0.82496-0 (2007).
Li X, Lin Z, Liu J, Tang Y, Yuan X, Li N, et al. Overall prevalence of human parvovirus B19 among blood donors in mainland China: A PRISMA-compliant meta-analysis. Medicine (Baltimore), 99:e19832. (2020) doi:https://doi.org/10.1097/MD.0000000000019832.
Jia, J. et al. Existence of various human parvovirus B19 genotypes in Chinese plasma pools: genotype 1, genotype 3, putative intergenotypic recombinant variants and new genotypes. Virol. J. 13, 155. https://doi.org/10.1186/s12985-016-0611-6 (2016).
Cheng, P., Jian, Q., Fu, Z. & Ma, Y. Parvovirus B19-associated severe anemia in adult liver transplant recipients: a case series and review of the literature. Surg. Infect. (Larchmt) 23, 848–856. https://doi.org/10.1089/sur.2022.186 (2022).
Huang, Q. et al. Parvovirus B19 infection in kidney transplant recipients: a prospective study in a teaching hospital in Shanghai, China. Transpl. Immunol. 74, 101667. https://doi.org/10.1016/j.trim.2022.101667 (2022).
Zhou, Y. et al. Detection of cytomegalovirus, human parvovirus B19, and herpes simplex virus-1/2 in women with first-trimester spontaneous abortions. J. Med. Virol. 87, 1749–1753. https://doi.org/10.1002/jmv.24218 (2015).
de Moraes, J. C. et al. Etiologies of rash and fever illnesses in Campinas Brazil. J. Infect. Dis. 204(Suppl 2), S627-636. https://doi.org/10.1093/infdis/jir490 (2011).
Toshev, A., Ivanova, S., Kovaleva, V., Andonova, L. & Mihneva, Z. Detection of Human parvovirus B19 (HPVB19) in serum samples from fever-rash ill individuals during the rubella outbreak (2005) in Bulgaria. Biotechnol. Biotechnol. Equip. 28, 1103–1107. https://doi.org/10.1080/13102818.2014.967746 (2014).
Qiu, Q. et al. Viral pathogenic spectrum and epidemiological features of rash and fever syndrome in Shanxi province, China, during 2009–2015. Chinese J. Virol. 33, 169–175. https://doi.org/10.13242/j.cnki.bingduxuebao.003117 (2017).
Wang, L. et al. The epidemiological characteristics of rash and fever syndrome in Gansu Province from 2009 to 2019. Chinese J. Dis. Control Prevent. 25, 1102–1106. https://doi.org/10.16462/j.cnki.zhjbkz.2021.09.019 (2021).
Ju, X., Xu, A., Fang, Q. & Huang, J. Etiology study on rash and fever illness in Guangdong Province, 2010–2012. Chinese J. Dis. Control and Prevent. 17, 670–673 (2013).
Fei, Y. et al. Study on the etiology of rash and fever illness in Pudong New Area of Shanghai from 2010 to 2017. Chinese J. Dis. Control and Prevent. 23, 550–554. https://doi.org/10.16462/j.cnki.zhjbkz.2019.05.012 (2019).
Chen M, Wang Y, Zhang Z, Zou L, Dou X, Huang F. Detection of rash and fever illness associated viruses in suspected measles cases in Bei**g from 2017 to 2018. Int. J. Virol. (2019) 26:
Nguyen, Q. T., Wong, S., Heegaard, E. D. & Brown, K. E. Identification and characterization of a second novel human erythrovirus variant, A6. Virology 301, 374–380. https://doi.org/10.1006/viro.2002.1585 (2002).
Schneider, B. et al. Simultaneous persistence of multiple genome variants of human parvovirus B19. J. Gen. Virol. 89, 164–176. https://doi.org/10.1099/vir.0.83053-0 (2008).
Toppinen, M. et al. The landscape of persistent human DNA viruses in femoral bone. Forensic. Sci. Int. Genet. 48, 102353. https://doi.org/10.1016/j.fsigen.2020.102353 (2020).
Gallinella, G. Parvovirus B19 Achievements and Challenges. ISRN Virol. 2013, 1–33. https://doi.org/10.5402/2013/898730 (2013).
Servant-Delmas, A., Lefrere, J. J., Morinet, F. & Pillet, S. Advances in human B19 erythrovirus biology. J. Virol. 84, 9658–9665. https://doi.org/10.1128/JVI.00684-10 (2010).
Jain, A. & Kant, R. Genotypes of erythrovirus B19, their geographical distribution & circulation in cases with various clinical manifestations. Indian J. Med. Res. 147, 239–247. https://doi.org/10.4103/ijmr.IJMR_1816_16 (2018).
Sanabani, S., Neto, W. K., Pereira, J. & Sabino, E. C. Sequence variability of human erythroviruses present in bone marrow of Brazilian patients with various parvovirus B19-related hematological symptoms. J. Clin. Microbiol. 44, 604–606. https://doi.org/10.1128/JCM.44.2.604-606.2006 (2006).
Alves, A. D. et al. Persistence of Parvovirus B19 in liver from transplanted patients with acute liver failure. Future Microbiol. 15, 307–317. https://doi.org/10.2217/fmb-2019-0224 (2020).
Garcia Sde, O. et al. Parvovirus among patients with cytopenia of unknown origin in Brazil: a case-control study. J. Clin. Microbiol. 49, 1578–1580. https://doi.org/10.1128/JCM.00077-11 (2011).
S. W, N.S. Y, K.E. B. Prevalence of parvovirus B19 in liver tissue: no association with fulminant hepatitis or hepatitis-associated aplastic anemia. J. Infect Dis.,187(10):1581-1586. (2003) doi:https://doi.org/10.1086/374781
Norja, P. et al. Bioportfolio: lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc. Natl. Acad. Sci. U S A 103, 7450–7453. https://doi.org/10.1073/pnas.0602259103 (2006).
Shackelton, L. A. & Holmes, E. C. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J. Virol. 80, 3666–3669. https://doi.org/10.1128/JVI.80.7.3666-3669.2006 (2006).
Stamenkovic, G. G. et al. Substitution rate and natural selection in parvovirus B19. Sci. Rep. 6, 35759. https://doi.org/10.1038/srep35759 (2016).
Seetha, D. et al. Molecular-genetic characterization of human parvovirus B19 prevalent in Kerala State India. Virol J. 18, 96. https://doi.org/10.1186/s12985-021-01569-1 (2021).
Musiani, M. et al. Immunoreactivity against linear epitopes of parvovirus B19 structural proteins. Immunodominance of the amino-terminal half of the unique region of VP1. J. Med. Virol. 60, 347–352 (2000).
Cui, A. et al. Development of a multiplex one-step real-time RT-PCR assay for the simultaneous detection of eight viruses associated with febrile rash illnesses. Biosafety and Health 2, 89–94. https://doi.org/10.1016/j.bsheal.2020.04.003 (2020).
Acknowledgements
We thank staff members of the FRI surveillance network laboratories and sentinel hospitals for assistance with field investigation, administration, data and specimen collection, and laboratory testing.
Funding
This work was supported by grants from the Key Technologies Research and Development Program of the National Ministry of Science (2009ZX10004202, 2013ZX10004202, 2018ZX10713002 and 2018ZX10713001-003).
Author information
Authors and Affiliations
Contributions
H.J. and A.C. prepared the manuscript; H.J., Q.Q., and Y.Z. performed the identification and sequencing; H.J., A.C., Y.Z. and X.L. analyzed the data; A.C., Q.Q. and H.J. designed the study; A.C. and W.X. acquired funding, and supervised the study. A.C. reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, H., Qiu, Q., Zhou, Y. et al. The epidemiological and genetic characteristics of human parvovirus B19 in patients with febrile rash illnesses in China. Sci Rep 13, 15913 (2023). https://doi.org/10.1038/s41598-023-43158-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43158-y
- Springer Nature Limited