
Abstract
In this study, the production of isomaltooligosaccharide from potato peel starch was carried out in three steps: liquefaction, saccharification, and transglucosylation. Further, cloning α-transglucosidase gene from Aspergillus niger (GH31 family), transforming into E. coli BL21 (DE3), overexpressing and purifying the resulting protein for the production of α-transglucosidase. The generated α-transglucosidase was then bound with magnetic nanoparticles, which improved reusability up to 5 cycles with more than 60% activity. All the modifications were characterized using the following methods: Fourier transform infra-red analysis, Transmission Electron Microscopy, Field Emission Scanning Electron Microscopy, Energy Dispersive X-ray spectroscopy, X-Ray Diffraction Spectroscopy, Thermogravimetric Analysis, and Dynamic Light Scattering (DLS) analysis. Further, the optimum conditions for transglucosylation were determined by RSM as follows: enzyme-to-substrate ratio 6.9 U g−1, reaction time 9 h, temperature 45 °C, and pH 5.5 with a yield of 70 g l−1 (± 2.1). MALDI-TOF–MS analysis showed DP of the IMOs in ranges of 2–10. The detailed structural characterization of isomaltooligosaccharide by GC–MS and NMR suggested the α-(1 → 4) and α-(1 → 6)-D-Glcp residues as major constituents along with minor α-(1 → 2) and α-(1 → 3) -D-Glcp residues.
Similar content being viewed by others


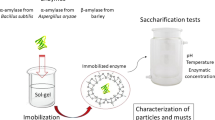
Introduction
Isomaltooligosaccharides (IMOs) are non-digestible but fermentable oligosaccharides that increase the growth of certain health-beneficial bacteria, particularly Bifidobacteria and lactobacilli, which influence gut metabolism and have an impact on gastrointestinal microbial ecology1,2,3,4,5,6. IMOs are prebiotic oligosaccharides composed of glucose units linked by mainly α-(1 → 6) and α-(1 → 4) glycosidic linkage with a lower proportion of α-(1 → 3) (nigerose) and α-(1 → 2) (kojibiose) glycosidic linkage7,8,9. Typically, IMOs have different degrees of polymerization (DP) which include isomaltose (DP2), isomaltotriose (DP3), isopanose, panose (DP3), isomaltotetrose (DP4), and isomaltopentose (DP5)10,11. The IMOs are not digested by human enzymes but fermented by gut flora which exerts their prebiotic effect5,12,13. In addition to the prebiotic effect, IMOs have a low glycemic index and act as a low-calorie sweetener that provides health benefits to diabetic persons10,14.
Commercially, IMOs are produced from starch taken from various sources (sweet potato, potato, tapioca, rice, and banana)11,15,16,17,18. Generally, the traditional method employed for the production of IMOs consisted of three steps: liquefaction, saccharification, and transglucosylation6,19. Recent studies have demonstrated the simultaneous saccharification and transglucosylation (SST) for the production of IMOs16,20. These studies improved the efficiency of IMOs production and decreased the reaction time. Different studies have also reported enzymatic production of IMOs from sucrose using dextransucrase and dextranase21,22,23. Further, it was reported that the development of magnetic nanoparticles (MNPs) based immobilization of α-transglucosidase is one best way to improve the reusability of the enzyme24,25,26.
In this study, the extraction of starch from potato peel and then liquefaction and saccharification of liquefied starch were first carried out. A gene encoding α-transglucosidase (average MW ~ 110 kDa) from Aspergillus niger (GH31 family) was synthesized by GenScript (Singapore) and cloned into the pET28a vector. Given the importance of enzymes for IMOs production, an effort was made to express heterologously in E. coli BL21(DE3). Further, the transglucosylation reaction was optimized by RSM to maximize the yield of IMOs. Thereafter, produced α-transglucosidase was immobilized with MNPs and characterized by various analytical techniques. Finally, the structural features of purified IMOs fraction were analysed by using MALDI-TOF–MS, GC–MS, and NMR.
Materials and methods
Materials
Potato peel waste was collected from the institute mess (NABI Mohali, Punjab, India) and used for starch extraction. All chemicals used in IMOs characterization and two enzymes, Termamyl® SC DS (A4862) and Fungamyl® (A8220), used for liquefaction and saccharification reaction were purchased by Sigma-Aldrich. The α-transglucosidase gene sequence of Aspergillus niger (GH31 family) was synthesized and cloned in the pET28a vector by GenScript Biotech Corporation. The standards glucose, maltose, maltotriose, isomaltose, isomaltotriose, panose, isopanose, and all other chemicals used in the present study were also purchased from Sigma-Aldrich.
Starch extraction
Potato peels (100 g) were first washed and then blended using a laboratory-scale blender. Immediately, the blended mixture was filtered and the left residue rinsed with deionized water (5 × 200 ml) 2 to 3 times. The filtrate was collected into a beaker and kept at 4 °C for settled-down starch. The supernatant was discarded and the white layer of starch was collected from the beaker into an oven tray and allowed to dry in a hot air drier at 37 °C for 24 h. Following this, dried residues were grounded in fine powder and stored in an airtight container for later use27,28.
Liquefaction
Effect of enzyme-to-substrate ratio
Extracted starch was added to water and made a slurry(1 g 10 ml-1). The pH of the slurry was fixed at 6.9 with lactic acid. Different ratios of an enzyme (Termamyl® SC DS) to substrate (starch) such as 0.3–5.5 U g-1 were used. The mixer was placed at 95 °C for 1 h. The residual starch was estimated with the iodine (KI/I2) test29.
Effect of pH
The starch slurry (1 g.10 ml-1) was adjusted at different pH (4–8). The slurry was liquefied with a fixed enzyme (Termamyl® SC DS) to substrate ratio (0.7 U g-1) at a fixed temperature of 95 °C for 1 h. The residual starch was estimated as mentioned above.
Effect of temperature
Initially, 1 g.10 ml-1 starch was hydrolysed with a fixed enzyme (Termamyl® SC DS) to substrate ratio (0.7 U g-1) and pH (6.9) at different temperatures (65–110 °C) for 1 h6,30,31.
Saccharification
Effect of enzyme-to-substrate ratio
The liquefied slurry was further saccharified with a different ratio of the enzyme (Fungamyl®) to substrate (0.7–9.6 U g-1) which affected the yield of maltooligosaccharide. The other three factors (temperature, pH, and time) were set at 50 °C, pH 5.5, and 12 h, respectively. After 12 h, the reaction was stopped, and TLC and HPAEC-PAD were used to analyse the reaction mixture6,30,31.
Effect of time
The effect of different reaction times (2–12 h) also affected the yield of maltooligosaccharide when the other three factors (enzyme to substrate ratio, temperature, and pH) were fixed at 1.2 U g-1, 50 °C, and pH 5.5, respectively. All reactions were stopped and analysed as mentioned above.
Effect of temperature
The liquefied slurry was saccharified with a fixed enzyme-to-substrate ratio (1.2 U g-1) and pH 5.5 at different temperatures (40–70 °C) for 4 h. After 4 h, reaction was stopped and analysed as mentioned above.
Effect of pH
The reaction mixture varied at different pH (4.5–7.5), affecting maltooligosaccharide yield when the other three factors (enzyme to substrate ratio, temperature, and time) were set at 1.2 U g-1, 50 °C, and 4 h, respectively. After 4 h, the reaction mixture was stopped by boiling mixture for 10 min and the reaction mixture was analysed as mentioned above.
Production of α-transglucosidase
The synthesized α-transglucosidase gene was cloned in pET-28a and transformed into E. coli BL21(DE3). The enzyme was induced with IPTG (0.2 mM) overnight at 18 °C in 1 L terrific broth. Afterward, cells were harvested by centrifugation (7000 rpm, 10 min) and lysed by sonication in buffer A (10 mM 4-(2-hydroxy-ethyl)-1-piperazineethanesulfonic acid [HEPES]) and cells were resuspended in an appropriate buffer having protease inhibitors as well as lysed by sonication. Subsequently, the supernatant was collected, the enzyme was purified by nickel-nitrilotriacetic acid (Ni–NTA) purification column, and molecular weight was determined by Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE)32.
Purification of α-transglucosidase
HisTrap HP column (GE Healthcare) was used to purify the α-transglucosidase protein. N’ terminal His6-tagged recombinant α-transglucosidase protein was put onto a column that had already been pre-equilibrated with buffer A (10 mM HEPES of pH 7.5, 250 mM NaCl). Buffer B (10 mM HEPES of pH 7.5, 250 mM NaCl, and 500 mM imidazole) was used to elute bound proteins in one step after washing with buffer A. Further α-transglucosidase protein was purified and concentrated with Amicon Ultra-15 100 kDa molecular weight cut-off concentrator. The purified α-transglucosidase was then kept at −80 °C for further usage32.
Secondary and Tertiary Structure characterization of α-transglucosidase protein
Circular dichroism (CD) spectroscopy was used to monitor the secondary structural alterations in free enzyme after immobilization. Biologic spectrometer MOS-500 equipment was used to record the UV CD spectra in the 190–280 nm wavelength region. The 0.2 mg/mL enzyme concentration was employed in all CD measurement experiments at 6 °C in a diluted buffer (Potassium phosphate buffer, pH 7.4). Tertiary structural model of α-transglucosidase was generated in Phyre 2 and processed in PyMOL software33. It was superimposed on a α-transglucosylase (protein data bank ID: 4B9Z) for identifying critical residues34.
Transglucosylation
Effect of enzyme-to-substrate ratio
The saccharified slurry was further treated by the 0.7–11 U g-1 enzyme (α-transglucosidase) to substrate (saccharified starch) ratio at three fixed factors, temperature, pH, and time were 45 °C, 5.5, and 12 h, respectively. The product was analysed by TLC and High-performance Anion Exchange Chromatography pulsed amperometric detection (HPAEC-PAD)1,2,3,4,5,6.
Effect of time
The saccharified starch was incubated with a 5.6 U g-1 enzyme (α-transglucosidase) to substrate (saccharified starch) ratio at 45 °C and pH 5.5. Samples were drawn at different time intervals (2–12 h) for time optimization. The product was analysed as mentioned above.
Effect of pH
The effect of pH on the IMOs yield varied between 3.5 and 7.5 when enzyme to substrate ratio, temperature, and time were set at 5.6 U g-1, 45 °C for 6 h in a shaking water bath, respectively. After 6 h, the reaction was stopped and the product was analysed by TLC and HPAEC- PAD.
Effect of temperature
The saccharified starch was placed at a different temperature varying between 35 and 75 °C and enzyme to substrate ratio, pH, and time were set at 5.6 U g-1, 5.5 pH for 6 h, respectively. The reaction mixture was analysed as mentioned above.
Experimental design for optimization of optimum condition for transglucosylation reaction and statistical analysis
The Box-Behnken was used to optimize the best condition for the transglucosylation reaction. The factors investigated in this study were enzyme-to-substrate ratio (A), slurry pH (B), temperature (°C), and reaction time (h). The three factors were evaluated at + 1, 0, and − 1 for high, intermediate, and low levels. The design contains a total of 29 runs based on Box-Behnken35.
α-transglucosidase immobilization
Synthesis of magnetic nanoparticles (Fe 3 O 4 )
The magnetic nanoparticles (Fe3O4) were synthesized by coprecipitation of ferric and ferrous chloride in the presence of 1.5 M NaOH as reductant. To prepare magnetic nanoparticles, solution 5.4 g FeCl3.6H2O was dissolved in 25 ml of 0.4 M HCl with 2 g FeCl2.4H2O and the reaction mixture was placed at 200 °C until a yellow transparent solution was formed. Further, the yellow transparent solution was dropwise added to 1.5 M NaOH solution at 80 °C. As the yellow solution was added to the NaOH solution, black precipitation of Fe3O4 formed. The precipitate was collected in a magnetic stand and washed with MQ water. The precipitate was stored in 200 ml of 0.1 M tetramethylammonium hydroxide (TMAOH) solution for further use24,26.
Synthesis of silica-coated magnetite nanoparticle Fe 3 O 4 @Si
Synthesized Fe3O4 was stabilized by silica coating, which also prevented agglomeration from forming as a result of interparticle interaction. TMAOH stored magnetite was put in a falcon magnetic stand, and the supernatant was discarded. Magnetite was transferred to the beaker with a 1:1.4 volume ratio of ethanol and 10% tetraethyl orthosilicate (TEOS). The mixture was placed at 90 °C for 6 h for the synthesis of silica-coated nanoparticles, followed by washing and storage24,26.
Functionalization of Fe 3 O 4 @Si with 16-Phosphonohexadecanoic acid (16-PHDA) linker
The obtained silica-coated magnetite nanoparticles were linked with a 16-PHDA linker in 1:1 ratio. The mixture was placed in an ultrasonicator for 30 min. After 30 min, the solution was rinsed with water and kept for future use24,26.
Immobilization of α-transglucosidase with 16-PHDA functionalized magnetite nanoparticle
In order to couple α-transglucosidase with the carboxylic groups of 16-PHDA, the mixture was first activated using EDC in 0.1 M MES buffer, then α-transglucosidase was added in an equal amount, and the mixture was left at room temperature for 2 h24. The Bradford method was used to calculate the amount of enzyme immobilized on nanoparticles in terms of protein contain. The following equation was used to determine the enzyme loading percentage:
Ei = Et–Es, Ei = Immobilized enzyme, Et = Initial amount of added enzyme, Es = Amount of enzyme in supernatant.
Characterization methods
Agilent Cary, 660 series with DTGS detector spectrophotometer, was used to record the IR spectra of the samples at frequencies between 400 and 4000 cm−1 26. Transmission electron microscopy with high resolution (HR-TEM, LIBRA 120), 300 kV used for the size and shape of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-16 PHDA, and Fe3O4@SiO2-16 PHDA-α-transglucosidase. The size and surface morphology of Fe3O4 and Fe3O4@SiO2 were examined using scanning microscopy (FE-SEM, Thermoscientific Apreo S)24,26. By using an X-ray diffractometer (XRD; Rigaku, Smart LAB SE), the purity and crystallinity of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-16 PHDA, and Fe3O4@SiO2-16-PHDA-α-transglucosidase were measured26,36. Further, the percentage weight loss of samples was analyzed by using thermogravimetric analysis (TGA) (Netzsch simultaneous thermal analyser STA 449F1) at temperatures ranging from 25 to 800 °C in a nitrogen (N2) atmosphere. Finally, the zeta potential and colloidal stability were measured using Dynamic Light Scattering (DLS) analysis24,26.
Stability and reusability of immobilized enzyme
To determine the stability of free and immobilized α-transglucosidase at different pH and temperature conditions, the maltooligosaccharides were incubated with α-transglucosidase at pH values ranging from 3.5 to 8.5 and temperatures (30–80 °C), respectively. Finally, the percent relative activity in each experiments represents the enzyme activity relative to the control, which was assumed to be 100%37,38.
The reusability of the immobilized α-transglucosidase was analyzed by hydrolysis of maltose, under optimized conditions (enzyme to substrate ratio 6.9 U g-1, reaction time 9 h, temperature 45 °C, and pH 5.5). After reaction completion, immobilized α-transglucosidase was recovered by a magnetic stand at the end of each cycle, thoroughly cleaned two to three times with deionized water, and then used for the subsequent cycle of the reaction. The recovered immobilized α-transglucosidase was reused in the fresh maltose solution. The first cycle of the immobilized enzyme's activity was considered the control, with 100% activity24.
Determination of kinetic parameters
The optimized assay condition was used to establish the kinetic parameters of free and immobilized α-transglucosidase, with the exception that the maltose concentration was changed from 100 to 500 mM. Michaelis–Menten and Lineweaver–Burk plots were used to calculate the kinetic parameters, including the Michaelis–Menten constant (Km), turnover number (Kcat), and catalytic efficiency (Kcat/Km)39.
Purification of IMOs
The crude IMOs were purified with HPLC-RI on a superdex peptide 10/300 column (10 × 300–310 mm). The sample was put in the chromatography cabinet at 40 °C. The autosampler was used to inject the sample (15 µl) into the column. The sample was eluted at a flow rate of 0.5 ml/min with deionized distilled water and the effluent was manually collected. Further, the purified sample was characterized by TLC, HPAEC-PAD, MALDI-TOF–MS, GC–MC, and NMR40.
Thin layer chromatography
IMOs samples (5 mg ml-1) were spotted at a distance of 1 cm from the bottom on TLC silica gel glass plates (60 F 254, 10 × 20 cm; TLC silica gel, Sigma Aldrich). After spotting samples were allowed to dry. The bottom part of the plate was submerged in the mobile phase (n-propanol/water/ethanol in 70:20:10 v/v/v). The sample spots drifted upward direction with the mobile phase. The plates were dried when the mobile phase reached the second end of the plate. The spraying solution (5gL-1α-naphthol and 50 mL L-1 H2SO4) was applied on the TLC plates. The plates were heated at 120 °C for 10 min18.
MALDI-TOF–MS
The MALDI-TOF–MS (AB SCIEX 5800) was used to record the mass spectrum of the purified IMOs sample. An equivalent volume of matrix solution (0.1 M 2,5-dihydroxybenzoic acid and 0.03 M 1-hydroxyisoquinoline in aq. 50 percent ACN) was mixed with an aqueous sample. The sample mixture was loaded on a MALDI target plate and dried41. A mass spectrum was generated by using the spectra from 200 laser pulses42.
Linkage analysis
To investigate the glycosyl linkage composition of IMOs, partially methylated alditol acetate derivatives were produced. The IMOs samples (5 mg) and 15 mg NaOH powder were added in dry DMSO. Each sample received 1 ml of iodomethane, and the mixtures were mixed for 30 min at room temperature. A nitrogen stream was used to remove excess iodomethane, and samples were divided into two layers: upper (water) and bottom (chloroform). The bottom layer (chloroform) was recovered and dried and stored for analysis. The derivatized samples were then analyzed using GC-FID-MS35,42.
NMR
The purified IMOs (10 mg) were dissolved in 0.5 ml D2O. A 600 MHz NMR spectrometer was used to generate 1H NMR spectra. Every 2D NMR spectrum was recorded with standard Bruker pulse techniques. The internal acetone standard was used to measure chemical changes41,42.
Statistical analysis
All the experiments were repeated in triplicate and analyzed by the Analysis of variance (ANOVA) test using Graph Pad 6.0 and Prism 543. The results of statistical analysis were expressed as mean ± SEM. Differences between the mean values of the measured properties were compared using multiple-range Tukey's test. In all cases, a p-value less than 0.05 was statistically significant44.
Results and discussion
Liquefaction and saccharification of starch extracted from potato peel
In the present study, starch from potato peel was used for IMOs production. Starch was isolated from wet potato peel with a yield of 6.5% (± 1.1). IMOs were produced using three enzymatic pathways: liquefaction, saccharification, and transglucosylation. In the first step, the optimum condition for nearly complete liquefaction of potato starch was determined as time: 1 h, temperature: 95 °C, pH: 6.9, and enzyme (Termamyl®) to substrate ratio: 0.7 U g-1. The completion of liquefaction was identified by the dispersion of color of the starch iodine complex during the reaction. Further, in the second step, Fungamyl® was used for the saccharification. The optimum condition for complete saccharification of liquefied starch was determined as time: 4 h, temperature: 50 °C, pH: 6–6.5, and enzyme to substrate ratio: 1.2 U g-1. The HPAEC-PAD analysis of the saccharified product showed the presence of maltose (72 g l-1), and maltotriose (18 g l-1) as major constituents along with a small amount of glucose (13 g l-1). The detailed explanation is provided in the supplementary data (Supplementary Fig. S1). The optimized condition of liquefaction and saccharification was found to be similar, as reported in the previous studies6,16.
Production of α-transglucosidase
The gene (α-transglucosidase) from A. niger was cloned in the pET-28a vector (GenScript Biotech Corporation) and further overexpressed. The purified α transglucosidase was analyzed on SDS-PAGE, indicating a molecular weight of about 110 kDa. The display image of SDS-PAGE (Fig. 1) cropped from the original image which is presented in Supplementary Fig. S2. In a previous study α-transglucosidase encoding gene from A. niger was cloned and expressed in Pichia pastoris to produce α-transglucosidase45. However, it was cloned and produced in E. coli in the current study for the first time.

10% SDS PAGE analysis for estimation of molecular weight of produced α- transglucosidase. (Lane 1 purified α-transglucosidase; lane 2 Crude enzyme (0.2 IPTG); lane 3 protein marker). The SDS PAGE display image cropped from the original image presented in Supplementary Fig. S2.
Secondary and tertiary structure analysis of α-transglucosidase
CD spectra of free and immobilized α-transglucosidase were studied to analyse the impact of immobilization on the enzyme's secondary structure. Overall, there are no changes in secondary structures of free and immobilized α-transglucosidase (Fig. 2a), suggesting that current immobilized technique does not interfere with the structure of α-transglucosidase which was also in agreement with the previous study34.

Secondary and tertiary analysis (a). CD spectrum of Immobilized magnetic nanoparticles (MNPs) with α-transglucosidase, and free α-transglucosidase; (b). Tertiary structural model of α-transglucosidase was generated in Phyre 2 and processed in PyMOL software. It was superimposed on a α-transglucosylase (protein data bank ID: 4B9Z) for identifying critical residues; (c). Crystal structure of α-transglucosylase (4B9Z) suggested that substrate-binding pocket of α-transglucosidase made up of narrow groove with residues of D371, I410, W488, H718, R644, W657, and D660.
The tertiary structure of α-transglucosidase (4B9Z) suggested that substrate-binding pocket of α-transglucosidase made up of narrow groove with residues of D371, I410, W488, H718, R644, W657, and D660 (Fig. 2b and c). In the current study, D371 and D660 are likely to act as nucleophilic attackers and acid–base residues of α-transglucosidase. These were revealed through superimposing a crystal structure of α-transglucosylase (4B9Z) on a model of α-transglucosylase whose D412 and D480 function as a nucleophilic attacker and an acid–base residue, respectively. The Asp660 is likely to catalyse maltooligosaccharide by breaking α-(1,4) glycosidic linkages and transferring glucose on maltose or glucose.
Optimization of α- transglucosylation of saccharified starch
In order to determine the optimum transglucosylation reaction for the production of IMOs, the effect of four variables enzyme-to-substrate ratio, temperature, pH, and time were investigated. The single-factor experimental data revealed that the highest yield of IMOs (58 g l-1) with glucose and maltose as other constituents were plotted by HPAEC-PAD data (supplementary Fig. S3) under the optimum condition such as enzyme-to-substrate ratio: 5.6 U g-1, temperature: 45 °C, pH: 5.5, and time 6 h1,2,3,4,5,6.
The Box-Behnken experimental design was employed to study optimum conditions to achieve the highest IMOs yield. The relationship between the independent and dependent variables can be expressed by yield: Y(IMOs) = 70.00–5.29A + 5.24.1B D-1.18 CD-5.76A2-2.23B2 + 0.72 C2-1.33D2. The IMOs yield data is shown in Table 1. ANOVA was used to determine the sufficiency and fitness of the impacts of independent factors on the yield (supplementary Table S1). The proposed model had a low P value (0.0001), showing a highly significant model. The adjusted R2 of determination, which was 0.9742, also indicated that this model was highly significant. The outcome showed that the IMOs yield was significantly affected by all independent (A, B, C) and quadratic factors (A2, B2, C2)35. The response surface plots in Fig. 3 showed the effects of independent variables and how they interacted with one another on the yield of IMOs. After transglucosylation the products were analyzed by HPAEC-PAD. The HPAEC-PAD estimated the highest yield of IMOs as 70 g l-1 (± 2.1) at a condition developed by the RSM model, which was enzyme to substrate ratio 6.9 U g-1, reaction time 9 h, temperature 45 °C, and pH 5.5.

Contour plots for the effect of variable parameters on the yield of IMOs, A. Reaction time and Enzyme/substrate ratio, B. Temperature and Enzyme/substrate ratio, C. pH and Enzyme/substrate ratio, D. Temperature and reaction time, E. pH and Reaction time, F. pH and Temperature.
Immobilization of α-transglucosidase
MNPs were used to immobilize the α-transglucosidase. FeCl2 and FeCl3 salts were co-precipitated in NaOH solution to make MNPs. The MNPs were then coated with 10% TEOS (Tetraethyl orthosilicate), functionalized via 16-PHDA, and immobilized with α-transglucosidase with high binding efficiency (82%)24.
Characterization
FT-IR analysis
FT-IR spectroscopy analysis data (Fig. 4) confirmed the formation of MNPs and the surface modifications. The functional group of FeT–O–FeO was observed around 600–550 cm−1 in MNPs. Due to the Si–O–Si bond, a stretching peak displayed around 1031.01–1080 cm−1, confirming the magnetite nanoparticles silica coating. The stretching peak was around 1100–900 cm−1 confirming P–O and P–O–Fe groups after 16-PHDA linking. The spectral signal at 1643.32 cm−1 confirmed the presence of the amide carbonyl (NH–CO) group of immobilized α-transglucosidase during immobilization. The FTIR results also agreed with the previously reported study24.

FTIR spectra of α-transglucosidase immobilized on MNPs. (a). Fe3O4; (b). Fe3O4@SiO2; (c). Fe3O4@SiO2-16-PHDA; (d). Fe3O4@SiO2-16-PHDA-α-transglucosidase.
TEM analysis
The size distribution and TEM images of the Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-16 PHDA, and Fe3O4@SiO2-16 PHDA-α-transglucosidase all support the quasi-spherical shape of the nanoparticles as showed in Fig. 5. The predicted diameter of uncoated Fe3O4 nanoparticles is around 20 nm which further increases to 35 nm following silica coating, as illustrated in Fig. 5. According to TEM data, nanoparticles that have been modified with a linker and an enzyme have little to no change in diameter, which also in agreement with previous study26. This could be because of their smaller electron density as organic molecules only provide inadequate TEM disparity.

TEM image of (a). Fe3O4, (b). Fe3O4@SiO2 (c). Fe3O4@SiO2-16-PHDA; (d). Fe3O4@SiO2-16-PHDA-α-transglucosidase (Scale bar 50 nm).
FESEM analysis
Field emission scanning electron microscopy investigated surface morphology and particle size/shape. The results showed in Fig. 6a, the average size of the sphere-shaped Fe3O4 particles was 20–25 nm. Further, Fe3O4 particles sphere size slightly increased about 34–38 nm after silica coating on the surface of the Fe3O4 particles (Fig. 6b), comparable with previous study26,46,47. There was no significant change in the physical appearance of the nanoparticles occurred following modification with 16-PHDA and α-trasglucosidase enzyme.

FESEM image of (a); Fe3O4 (b). Fe3O4@SiO2; EDX image of (c). Fe3O4 and (d). Fe3O4@SiO2.
EDX analysis
The energy dispersive X-ray (EDX) spectroscopy results were used to characterize the mass percent composition of the synthesized Fe3O4 and Fe3O4 /SiO2 particles. Figure 6c showed Fe3O4 had a mass percent composition of 64.8% Fe, 31.4% O, and 3.9% C. After silica coating, the atomic composition of the nanospheres were Fe 63.0%, O 23.6%, C 8.9%, and Si 3.7% as depicted in Fig. 6d. The overall results were in agreement with previous study46.
XRD analysis
X-Ray Diffraction Spectroscopy (XRD) analysis was performed to confirm the purity and crystallinity of Fe3O4, Fe3O4@SiO2, organophosphorus linker, and enzyme-modified nanoparticles. Figure 7 showed the magnetite nanoparticles diffractogram peaks at 2θ = 30.2°, 35.6°, 43.2°, 57.3° and 62.9° attributed to (220), (311), (400), (422), (511) and (440) reflections, demonstrating the acceptable crystallinity of nanoparticles which was comparable with previously published data26,36. The overall result suggested that MNPs did not change crystalline structure after enzyme immobilization.

XRD spectra of (a). Fe3O4 (b). Fe3O4@SiO2 (c). Fe3O4@SiO2-16-PHDA.
TGA analysis
TGA analysis was performed to determine the percentage weight loss of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-16 PHDA, and Fe3O4@SiO2-16 PHDA-α-transglucosidase (Fig. 8). The samples were heated to a range of 25–800 °C to analyse the percentage loss of weight. At temperatures below 200 °C, all samples exhibited a minor initial mass loss due to the desorption of adsorbed water. As the temperature increased above 200–500 °C, weight loss was not observed in Fe3O4 nanoparticles. However, weight losses of about 3.2%, 8.8.%, and 12.0% were recorded in Fe3O4@SiO2, Fe3O4@SiO2-16-PHDA, and Fe3O4@SiO2-16-PHDA-α-transglucosidase respectively which were also in accordance to previously published data36. The Additional loss of around 3.2% confirmed the successful binding of α-transglucosidase on MNPs.

Thermogravimetric analysis (TGA) of (a). Fe3O4 (b). Fe3O4@SiO2 (c). Fe3O4@SiO2-16-PHDA (d). α-transglucosidase bound Fe3O4@SiO2-16-PHDA.
DLS analysis
Zeta potential was used to assess how the surface charge of the enzyme and MNPs in the solution affected the stability of its colloidal particles. The stability of the colloidal solution would increase with increasing zeta potential. The results of measuring the zeta potential of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-16-PHDA, and Fe3O4@SiO2-16-PHDA-α-transglucosidase are showed in Table 2. The zeta potential of Fe3O4@SiO2-16-PHDA increased to positive levels (from −46.0 to −31.0) after α-transglucosidase immobilization, indicating that less negative α-transglucosidase neutralized the negatively charged MNPs which also in agreement with the previous study24. Further the PDI values of all the MNPs in the range of 0.161–0.212 suggested higher colloidal stability.
Stability and Reusability of immobilized α-transglucosidase
Free and immobilized α-transglucosidase activities were measured in the temperature and pH range of 30–80 °C and 3.5–8.5, respectively. The results revealed that the optimum temperature for free α-transglucosidase was 45 °C which was slightly pushed towards 55 °C for immobilized α-transglucosidase (Fig. 9a). Further, significant reduction (30% at 80 °C) in the activity of the free α-transglucosidase whereas the immobilized enzyme retained nearly 70% activity at 80 °C. Figure 9b revealed that free and immobilized α-transglucosidase exhibited nearly similar activity at optimum pH 5.5. However, higher retention in the activity was observed for immobilized α-transglucosidase compared to the free enzyme at pH 8.5. The overall result was also in agreement with previous studies suggesting that magnetic nanoparticle immobilization improved the thermal and pH stability37.

Stability of free and immobilized α-transglucosidase (a). at different temperature range 30–80 °C and (b). pH range 3.5–8.5. (c). Reusability of immobilized α-transglucosidase in subsequent 10 cycles of reaction conducted at optimized conditions (6.9 U g-1, 45 °C, 9 h and pH 5.5). The first cycle enzyme activity considered as 100%. All data are represented as mean ± SD (n = 3).
The advantage of MNPs is easy removal from a reaction mixture by using simple magnet which provide magnetic nanoparticle stand-alone-candidate to other nonmagnetic nanoparticles. The result showed in Fig. 9c clearly demonstrated that immobilized α-transglucosidase retained its activity up to more than 60% after 5 reaction cycles. Further, it was observed that the maximum yield of IMOs (64 g l-1 ± 2.3) was achieved with immobilized α-transglucosidase at the optimum condition as enzyme to substrate ratio 6.9 U g-1, reaction time 9 h, reaction temperature 45 °C, and pH 5.524.
Analysis of enzyme kinetics parameter of α-transglucosidase
The analysis of enzyme kinetic parameters of free and immobilized α-transglucosidase such as, Michaelis–Menten constant (Km), turnover number (Kcat), and catalytic efficiency (Kcat/Km), was evaluated using a GraphPad Prism software version 543. The enzymatic parameters were calculated using Michaelis–Menten and Lineweaver–Burk plots. Fig. S4 illustrates the outcomes of the α-transglucosidase kinetic study at different maltose concentrations. The result showed up to 300 mM maltose, the reaction rate and maltose concentration are correlated linearly. The reaction rate was stabilized when the concentration of maltose was larger than 300 mM. The results showed in Table 3 support the findings in Fig. S4 in their observations. The Km values of free and immobilized enzymes were 97.85 ± 11.40 and 81.08 ± 9.990, respectively. The best-fit value of Kcat is 1107 ± 14.28 and 715.6 ± 9.528 for free and immobilized enzymes39.
Thin layer chromatography and HPAEC-PAD analysis
TLC and HPLC were used to analyse the transglucosylation reaction products (Fig. S5). As the hydrolysis time increased from Lane 1 (6 h) to Lane 5 (30 h), glucose concentration increased, and oligosaccharide concentration decreased, as showed in Fig. S5a. This is due to the fact that oligosaccharides degrade into glucose as time goes on, making the glucose band darker over time and the oligosaccharide band disappear. Fig. S5b showed HPAEC-PAD chromatogram of transglucosylation reaction products after 6 h hydrolysis time18.
Size-exclusion chromatography purification and MALDI-TOF–MS analysis of IMOs
The product obtained after transglucosylation of saccharified starch was separated by size-exclusion chromatography. Further, HPAEC-PAD analysis revealed the glucose content in the purified IMOs rich fraction was reduced to 4 g l-1 (Supplementary data Fig. S6). The other constituents in the purified fractions were to be as; isomaltose: 37.7 g l-1, isomaltotriose: 21.6 g l-1, panose: 17 g l-1, maltose: 3 g l-1, and another oligosaccharide: 20 g l-1. The purity of the IMOs rich fraction was nearly 85% which was nearly similar as described in the previous study48.
MALDI-TOF–MS was used to analyze the purified IMOs. The analysis showed the presence of oligosaccharides DP 2–10 (m/z 365–1337 Da) showed in Fig. 10, comparable to previously published data49. However, according to m/z peak analysis, the amount of oligosaccharides DP > 10 was very low and undetectable.

MALDI-TOF–MS spectroscopy of the formed IMOs generated after the treatment of saccharified starch with α-transglucosidase.
Methylation analysis of IMOs oligosaccharides
The glycosyl linkage analysis of IMOs indicates that terminal glucopyranosyl (Glcp), α-(1 → 4), and α-(1 → 6) D linked Glcp residues were more abundant in the molar ratio of nearly 1.02:0.43:1.0 with the small amount of α-(1 → 2) and α-(1 → 3)—linked Glcp residues in the relative molar ratio of nearly 0.12 and 0.05. The linkage analysis data is shown in Table 4, and detailed figures are provided in the supplementary data (Supplementary Fig. S7). The study further indicated that IMOs contain α-(1 → 4) and α-(1 → 6)- linked Glcp residues as the primary linkage present in isomaltose, isomaltotriose, and panose which was comparable with previously reported study50.
NMR analysis
The purified IMOs in which fraction was characterized by 1H, 13C, and 2D NMR (TOCSY, and HSQC) NMR spectroscopy (supplementary Fig. S8).
1H NMR spectroscopy showed the anomeric signal at δ 5.225(α) and δ 4.649 (β) in arrangement with a reducing α-(1 → 4) D Glcp unit. Further, the groups of anomeric signals along δ 5.389–5.395 and 4.970 revealed the presence of α-(1 → 4) and α-(1 → 6) linkage, respectively. The α-(1 → 6): α-(1 → 4) linkage ratio was found to be about 1.0:0.67, which suggested the presence of significantly higher α-(1 → 6) linkages. The result was also in agreement with the ratio of α-(1 → 6): α-(1 → 4) linkage obtained by glycosyl linkage analysis.
13C chemical shift was deduced from 13C and HSQC measurements. The analysis revealed the presence of anomeric signals of α-(1 → 4) and α-(1 → 6) linkage in the range of δc 100.7 and 98.3, respectively. The anomeric signals of δc 92.6 and 96.6 were indicative of reducing α-(1 → 4) D-Glcp and β-(1 → 4) residues, respectively. The anomeric signals agreed with previously reported results11. The anomeric signal of δH/δC at 5.427/90.2 and 4.785/97.1 suggested the presence of reducing α-(1 → 2) D-Glcp and β-(1 → 2) D-Glcp residues. Furthermore, the anomeric signal at δH/δC at 5.233/93.2 and 4.663/96.8 were indicative of reducing α-(1 → 3) D-Glcp and β-(1 → 3) residues. The detailed NMR analysis is summarized in Table 5.
Conclusions
IMOs have been produced by potato peel starch through three-step liquefaction, saccharification, and transglucosylation. A recombinant α-transglucosidase from A. Niger has been produced in E. coli. The α-transglucosidase was immobilized with MNPs for reusability (5 cycles more than 60% activity) and characterized by using FT-IR, TEM, FESEM, EDX, XRD, TGA, and DLS analysis. The maximum IMOs yield (70 g l-1) was achieved by RSM. The detailed structural characterization of IMOs suggested DP in ranges of 2–10 with the presence of α- (1 → 4) and α- (1 → 6) -D-Glcp residues as major constituents.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Thitaram, S. N. et al. Isomaltooligosaccharide increases cecal Bifidobacterium population in young broiler chickens1. Poult. Sci. 84, 998–1003. https://doi.org/10.1093/ps/84.7.998 (2005).
Zhang, L. et al. Effects of dietary isomaltooligosaccharide levels on the gut microbiota, immune function of sows, and the diarrhea rate of their offspring. Front. Microbiol. https://doi.org/10.3389/fmicb.2020.588986 (2021).
Hu, Y., Ketabi, A., Buchko, A. & Gänzle, M. G. Metabolism of isomalto-oligosaccharides by Lactobacillus reuteri and bifidobacteria. Lett. Appl. Microbiol. 57, 108–114. https://doi.org/10.1111/lam.12076 (2013).
Kondepudi, K. K., Ambalam, P., Nilsson, I., Wadström, T. & Ljungh, Å. Prebiotic-non-digestible oligosaccharides preference of probiotic bifidobacteria and antimicrobial activity against Clostridium difficile. Anaerobe 18, 489–497. https://doi.org/10.1016/j.anaerobe.2012.08.005 (2012).
Wu, Q. et al. Fermentation properties of isomaltooligosaccharides are affected by human fecal enterotypes. Anaerobe 48, 206–214. https://doi.org/10.1016/j.anaerobe.2017.08.016 (2017).
Cui, J. et al. Production, purification and analysis of the isomalto-oligosaccharides from Chinese chestnut (Castanea mollissima Blume) and the prebiotics effects of them on proliferation of Lactobacillus. Food Bioprod. Process. 106, 75–81. https://doi.org/10.1016/j.fbp.2017.08.003 (2017).
Cho, S. K. et al. An improved process of isomaltooligosaccharide production in kimchi involving the addition of a Leuconostoc starter and sugars. Int. J. Food Microbiol. 170, 61–64 (2014).
Zeng, M. et al. Facile and efficient chemical synthesis of gluco-oligosaccharides (GlcOS) with diverse glycosidic linkages as potential prebiotics to promote the growth of probiotic bacteria. Food Res. Int. 165, 112436. https://doi.org/10.1016/j.foodres.2022.112436 (2023).
Hu, Y., Winter, V. & Gänzle, M. In vitro digestibility of commercial and experimental isomalto-oligosaccharides. Food Res. Int. 134, 109250. https://doi.org/10.1016/j.foodres.2020.109250 (2020).
Sorndech, W., Nakorn, K. N., Tongta, S. & Blennow, A. Isomalto-oligosaccharides: Recent insights in production technology and their use for food and medical applications. LWT 95, 135–142. https://doi.org/10.1016/j.lwt.2018.04.098 (2018).
Kaulpiboon, J., Rudeekulthamrong, P., Watanasatitarpa, S., Ito, K. & Pongsawasdi, P. Synthesis of long-chain isomaltooligosaccharides from tapioca starch and an in vitro investigation of their prebiotic properties. J. Mol. Catal. B Enzym. 120, 127–135. https://doi.org/10.1016/j.molcatb.2015.07.004 (2015).
Zhang, N. et al. Functional oligosaccharide fermentation in the gut: Improving intestinal health and its determinant factors-a review. Carbohydr. Polym. 284, 119043–119116. https://doi.org/10.1016/j.carbpol.2021.119043 (2022).
Palframan, R. J., Gibson, G. R. & Rastall, R. A. Effect of pH and dose on the growth of gut bacteria on prebiotic carbohydrates in vitro. Anaerobe 8, 287–292. https://doi.org/10.1006/anae.2002.0434 (2002).
Himat, A. S. et al. Starch-based novel ingredients for low glycemic food formulation. Bioactive Carbohydr. Dietary Fibre 26, 100275. https://doi.org/10.1016/j.bcdf.2021.100275 (2021).
Duong Hong, Q. et al. Synthesis of isomaltooligosaccharides (IMOs) from sweet potato starch by simultaneous saccharification and transglycosylation using Saccharomyces cerevisiae Var. diastaticus BE 134 to improve purity of IMOs. J. Food Quality 2021, 1987219–1987316. https://doi.org/10.1155/2021/1987219 (2021).
Basu, A., Mutturi, S. & Prapulla, S. G. Production of isomaltooligosaccharides (IMO) using simultaneous saccharification and transglucosylation from starch and sustainable sources. Process Biochem. 51, 1464–1471. https://doi.org/10.1016/j.procbio.2016.06.008 (2016).
Plongbunjong, V., Graidist, P., Knudsen, K. E. B. & Wichienchot, S. Isomaltooligosaccharide synthesised from rice starch and its prebiotic properties in vitro. Int. J. Food Sci. Technol. 52, 2589–2595. https://doi.org/10.1111/ijfs.13545 (2017).
Chockchaisawasdee, S. & Poosaran, N. Production of isomaltooligosaccharides from banana flour. J. Sci. Food Agric. 93, 180–186. https://doi.org/10.1002/jsfa.5747 (2013).
Gómez, J. A. et al. Towards a biorefinery processing waste from plantain agro–Industry: Process development for the production of an isomalto–oligosaccharide syrup from rejected unripe plantain fruits. Food Bioprod. Process. 133, 100–118. https://doi.org/10.1016/j.fbp.2022.03.005 (2022).
Niu, D. et al. Highly efficient enzymatic preparation of isomalto-oligosaccharides from starch using an enzyme cocktail. Electron. J. Biotechnol. 26, 46–51 (2017).
Goulas, A. K., Fisher, D. A., Grimble, G. K., Grandison, A. S. & Rastall, R. A. Synthesis of isomaltooligosaccharides and oligodextrans by the combined use of dextransucrase and dextranase. Enzyme Microb. Technol. 35, 327–338. https://doi.org/10.1016/j.enzmictec.2004.05.008 (2004).
Kim, Y.-M., Seo, M.-Y., Kang, H.-K., Atsuo, K. & Kim, D. Construction of a fusion enzyme of dextransucrase and dextranase: Application for one-step synthesis of isomalto-oligosaccharides. Enzyme Microb. Technol. 44, 159–164. https://doi.org/10.1016/j.enzmictec.2008.10.007 (2009).
Huang, S.-X. et al. Enzymatic synthesis of non-digestible oligosaccharide catalyzed by dextransucrase and dextranase from maltose acceptor reaction. Biochem. Biophys. Res. Commun. 523, 651–657. https://doi.org/10.1016/j.bbrc.2019.12.010 (2020).
Singh, V. et al. Metallic/bimetallic magnetic nanoparticle functionalization for immobilization of α-amylase for enhanced reusability in bio-catalytic processes. Biores. Technol. 214, 528–533. https://doi.org/10.1016/j.biortech.2016.05.002 (2016).
Kumari, A. et al. Multiple thermostable enzyme hydrolases on magnetic nanoparticles: An immobilized enzyme-mediated approach to saccharification through simultaneous xylanase, cellulase and amylolytic glucanotransferase action. Int. J. Biol. Macromol. 120, 1650–1658 (2018).
Singh, V. et al. Xylanase immobilization on magnetite and magnetite core/shell nanocomposites using two different flexible alkyl length organophosphonates: Linker length and shell effect on enzyme catalytic activity. Int. J. Biol. Macromol. 115, 590–599 (2018).
Yusuph, M., Tester, R. F., Ansell, R. & Snape, C. E. Composition and properties of starches extracted from tubers of different potato varieties grown under the same environmental conditions. Food Chem. 82, 283–289. https://doi.org/10.1016/S0308-8146(02)00549-6 (2003).
Bergthaller, W., Witt, W. & Goldau, H.-P. Potato starch technology. Starch–Stärke 51, 235–242. https://doi.org/10.1002/(SICI)1521-379X(199907)51:7%3c235::AID-STAR235%3e3.0.CO;2-7 (1999).
Halick, J. V. & Keneaster, K. The use of a starch iodine-blue test as a quality indicator of white milled rice. Cereal Chem. 33, 315–319 (1956).
Wang, P. et al. Comparison of raw starch hydrolyzing enzyme with conventional liquefaction and saccharification enzymes in dry-grind corn processing. Cereal Chem. 84, 10–14. https://doi.org/10.1094/CCHEM-84-1-0010 (2007).
Komaki, T. & Taji, N. Studies on enzymatic liquefaction and saccharification of starch. Agric. Biol. Chem. 32, 860–872. https://doi.org/10.1080/00021369.1968.10859147 (1968).
Kuhaudomlarp, S., Stevenson, C. E. M., Lawson, D. M. & Field, R. A. The structure of a GH149 β-(1 → 3) glucan phosphorylase reveals a new surface oligosaccharide binding site and additional domains that are absent in the disaccharide-specific GH94 glucose-β-(1 → 3)-glucose (laminaribiose) phosphorylase. Proteins: Struct. Funct. Bioinform. 87, 885–892. https://doi.org/10.1002/prot.25745 (2019).
Bramucci, E., Paiardini, A., Bossa, F. & Pascarella, S. PyMod: sequence similarity searches, multiple sequence-structure alignments, and homology modeling within PyMOL. BMC Bioinform. 13, 1–6 (2012).
Zhang, L. et al. Laccase-immobilized tannic acid-mediated surface modification of halloysite nanotubes for efficient bisphenol-A degradation. RSC Adv. 9, 38935–38942 (2019).
Bijalwan, V., Ali, U., Kesarwani, A. K., Yadav, K. & Mazumder, K. Hydroxycinnamic acid bound arabinoxylans from millet brans-structural features and antioxidant activity. Int. J. Biol. Macromol. 88, 296–305 (2016).
Sahu, A., Badhe, P. S., Adivarekar, R., Ladole, M. R. & Pandit, A. B. Synthesis of glycinamides using protease immobilized magnetic nanoparticles. Biotechnol. Rep. 12, 13–25. https://doi.org/10.1016/j.btre.2016.07.002 (2016).
Patel, S. N. et al. Development of a thermo-stable and recyclable magnetic nanobiocatalyst for bioprocessing of fruit processing residues and D-allulose synthesis. Biores. Technol. 247, 633–639 (2018).
Hosseinipour, S. L., Khiabani, M. S., Hamishehkar, H. & Salehi, R. Enhanced stability and catalytic activity of immobilized α-amylase on modified Fe 3 O 4 nanoparticles for potential application in food industries. J. Nanopart. Res. 17, 1–13 (2015).
McCleary, B. V. et al. Purification, properties, and industrial significance of transglucosidase from Aspergillus niger. Carbohyd. Res. 185, 147–162 (1989).
Richter, A. et al. Preparation of starch and soluble sugars of plant material for the analysis of carbon isotope composition: A comparison of methods. Rapid. Commun. Mass Spectrom: An Int. J. Devoted Rapid Dissem. Up-to-the-Minute Res. Mass Spectrom. 23, 2476–2488 (2009).
Mazumder, K. & York, W. S. Structural analysis of arabinoxylans isolated from ball-milled switchgrass biomass. Carbohyd. Res. 345, 2183–2193. https://doi.org/10.1016/j.carres.2010.07.034 (2010).
Ali, U., Kanwar, S., Yadav, K., Basu, S. & Mazumder, K. Effect of arabinoxylan and β-glucan stearic acid ester coatings on post-harvest quality of apple (Royal Delicious). Carbohyd. Polym. 209, 338–349. https://doi.org/10.1016/j.carbpol.2019.01.002 (2019).
Motulsky, H. Prism 5 statistics guide, 2007. GraphPad Soft. 31, 39–42 (2007).
Kanwar, S., Ali, U. & Mazumder, K. Effect of cellulose and starch fatty acid esters addition on microstructure and physical properties of arabinoxylan films. Carbohyd. Polym. 270, 118317 (2021).
Liu, X., Wu, D., Wu, J. & Chen, J. Optimization of the production of Aspergillus niger α-glucosidase expressed in Pichia pastoris. World J. Microbiol. Biotechnol. 29, 533–540. https://doi.org/10.1007/s11274-012-1207-y (2013).
Rehman, G. U., Ismail, A., Goh, P., Arzhandi, M. R.-D. & Ismail, N. Aptes and teos modified binary recyclable hybrid Fe3O4@ GO nanocomposite for photocatalytic dye removal. Jurnal Teknologi 80 (2018).
Zeynizadeh, B. & Gilanizadeh, M. Green and highly efficient approach for the reductive coupling of nitroarenes to azoxyarenes using the new mesoporous Fe 3 O 4@ SiO 2@ Co–Zr–Sb catalyst. Res. Chem. Intermed. 46, 2969–2984 (2020).
Rengarajan, S. & Palanivel, R. High purity prebiotic isomalto-oligosaccharides production by cell associated transglucosidase of isolated strain Debaryomyces hansenii SCY204 and selective fermentation by Saccharomyces cerevisiae SYI065. Process Biochem. 98, 93–105 (2020).
Sorndech, W. Isomaltooligosaccharides as Prebiotics and their Health Benefits. Probiotics, Prebiotics and Synbiotics: Technological Advancements Towards Safety and Industrial Applications, 361–377 (2022).
Ota, M., Okamoto, T., Hoshino, W. & Wakabayashi, H. Action of α-d-glucosidase from Aspergillus niger towards dextrin and starch. Carbohyd. Polym. 78, 287–291. https://doi.org/10.1016/j.carbpol.2009.03.047 (2009).
Acknowledgements
The authors express gratitude to NABI Executive Director for his encouragement and assistance. The National Agri-Food Biotechnology Institute (NABI) and the University Grants Commission (UGC) are thanked by the authors for their financial assistance. For the NMR experiments, the authors are grateful to the IISER Mohali NMR research facility. We appreciate Dr. Kanti Kiran Kondepudi for help in research work and Atul Kesarwani's technical assistance. We also appreciate Raminder Kaur for technical assistance in recording CD spectra.
Author information
Authors and Affiliations
Contributions
Authors contributions Rohit Maurya contributed (Extraction of starch; Enzyme production, purification, immobilization and characterization; IMOs production, RSM optimization, purification and characterization): investigation; methodology; formal analysis; writing original draft. Usman Ali contributed (RSM optimization of IMOs): investigation; methodology; formal analysis. Sunaina Kaul contributed (Enzyme immobilization): investigation; methodology; formal analysis. Raja Bhaiyya contributed (Enzyme production): investigation; methodology; formal analysis. Ravindra Pal Singh: supervision; methodology; writing-review and editing. Koushik Mazumder: conceptualization; supervision; project administration; methodology; writing-original draft; writing-review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maurya, R., Ali, U., Kaul, S. et al. Immobilization of α-transglucosidase on silica-coated magnetic nanoparticles and its application for production of isomaltooligosaccharide from the potato peel. Sci Rep 13, 12708 (2023). https://doi.org/10.1038/s41598-023-38266-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38266-8
- Springer Nature Limited