
Abstract
Oral squamous cell carcinoma (OSCC) in the context of oral submucous fibrosis (OSF) has a high incidence owing to undefined pathogenesis. Identifying key genes and exploring the underlying molecular mechanisms involved in the conversion of OSF into OSCC are in urgent need. Differentially expressed genes (DEGs) between OSCC and OSF were dug from GEO databases and a total of 170 DEGs were acquired. Functional association of DEGs were analyzed by GO and KEGG. Protein–protein interactions (PPIs) analysis was carried out and candidate biomarkers were identified by Gene co-expression analysis and Cox analyses. Hub genes were confirmed by qRT-PCR in tissues and cell lines, of which we found that IRF4 mRNA was successively up-regulated from Normal to OSF and then to OSCC and associated with immune infiltrating levels. In addition, Immunohistochemical (IHC) and Immunofluorescence (IF) assays were conducted to validate the consistent upregulation of IRF4 and the oncogene role of IRF4 in OSF and OSCC at translation level. IRF4 may be indicative biomarker in transformation of OSF into OSCC. High IRF4 expression contribute to increased immune infiltration of OSCC and may provide a novel diagnostic marker for OSCC patients translated from OSF.
Similar content being viewed by others

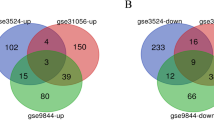
Introduction
It is estimated that 10–20% of the world's population is accustomed to chewing betel nut. Epidemiological study results suggested that betel nut is the main cause of oral submucosal fibrosis (OSF), and consumption of betel nut is also associated with oral squamous cell carcinoma (OSCC)1,2. OSF is a precancerous condition with a propensity for malignant transformation and up to a quarter of cases are present with epithelial dysplasia at biopsy. Malignant transformation rates have been estimated to range from 5.6 to 9.13 percent in recent studies3,4. OSCC is the most common malignant tumor of the head and neck5, and also the sixth highest incidence of cancer worldwide6,7. It is further observed that OSCC originating from OSF tends to occur in young adults, commonly in the posterior buccal, gingival and vestibular mucosa, and are more clinically aggressive and metastatic8. The prognosis and clinicopathological features of OSCC patients with OSF are inferior to conventional OSCC patients9, so it is urgent to search for new OSCC diagnostic biomarkers in the context of OSF.
The role of tumor immunology in tumorigenesis and progression is significant. Numerous types of cancer form ectopic lymphoid aggregates, also called tertiary lymphoid structures (TLSs), which are relevant to superior prognosis and response to immunotherapy10. The feature of tumor microenvironment (TME) is nutrition competition or coordination between tumor and infiltrating immune cells that influences antitumor immunity. Adaptive immune responses are essential for the clearance of tumors11. In the aspect of OSCC, inflammatory mediators are identified as potential markers for diagnosis and prognosis of OSCC12. The tumor microenvironment impacts evasion of OSCC from immune recognition and destruction13.
As a member of the IRF transcription factor family, interferon regulatory factor 4 (IRF4) is expressed and crucial for the development and function of numerous immunocyte types such as B cell, T cell and dendritic cell. IRF4 plays a significant role in autoimmune diseases14. In various mature lymphoid neoplasms, abnormally expressed IRF4 also acts as an oncogene. IRF4 and its upstream factor NF-κB form a regulatory circuit to promote the oncogenic transcriptional program in malignant lymphoid cells15. However, research on the role of IRF4 in OSCC is still lacking16.
In this study, we conducted a comprehensive bioinformatics analysis including functional enrichment analysis, CNV and immune infiltration analysis through Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases to identify key genes and explore the underlying molecular mechanisms involved in OSF into OSCC. We finally found the role of IRF4 in predictabilities of the transformation from OSF to the presence of OSCC. Understanding of the potential oncogenic axis may enable the discovery of noninvasive disease-specific diagnostic biomarkers. There is a discovery that IRF4 can be employed as a possible diagnostic and immunological predictor of malignant transformation of OSF into OSCC. This study may broaden the application of IRF4 in immunotherapy.
Materials and methods
Cell culture
Two human cell lines (HOK, HN4) were obtained from ATCC. The HN4 (OSCC cell line) and human oral keratinocytes (HOK, Normal control) cell line were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (Gibco, C11995500BT) and 1% penicillin/streptomycin (NCM Biotech, C125C5). Cells were incubated at 37 °C with 5% CO2.
Clinical samples
This study was approved by the ethical committee department of Affiliated Hospital of Stomatology of Nan**g Medical University (PJ2022-086-001). Research was performed in accordance with relevant guidelines and regulations. Informed consent was obtained from all patients. A total of 8 Normal oral mucosa tissues (Normal) and 10 oral squamous cell carcinoma (OSCC) samples were obtained in Stomatological College of Nan**g Medical University. The normal oral mucosa tissues were collected from the wounds of patients from whom impacted third molar were extracted. 10 oral submucosal fibrosis (OSF) samples were collected at Hunan ** DEGs. (B) KEGG pathways analysis of DEGs. Size of the dots represent Gene Counts, color of the dots are defined by the p-adjusted value. (C, D) The top 10 GO terms and KEGG pathways of GSE64216 and GSE23558 from GSEA analysis. (E) The PPI network of DEGs using the STRING database. Up- and down-regulated genes were marked in red and blue respectively.
In addition, Gene set enrichment analysis (GSEA) analysis was conducted using GSEA software, which showed that “DEFENSE RESPONSE TO SYMBIONT, DNA DEPENDENT DNA REPLICATION, MITOTIC NUCLEAR DIVISION, etc.” GO terms (Fig. 2C) and “ARACHIDONIC ACID METABOLISM, CELL CYCLE, DNA REPLICATION, etc.” KEGG pathways were significant in OSCC compared with OSF and Normal samples (Fig. 2D). GSEA analysis of 858 DEGs in GSE64216 and 3338 DEGs in GSE23558 datasets were displayed in supplementary Fig. S1A and S1B respectively.
Co-expression analysis of these 170 DEGs were performed via construction PPI network, and the results was visualized through cytoscape. 5 protein coding gene clusters were obtained, of which the cluster centered around CFTR and CXCL12 may play a critical role in transformation of OSF into OSCC (Fig. 2E).
Screening of hub genes assisted with TCGA database
The Cancer Genome Atlas (TCGA) OSCC cohort including 505 OSCC tissues and 24 Normal tissues was used to further screen hub genes (Fig. 3A). GSEA analysis displayed enriched GO terms and KEGG pathways in TCGA data (Fig. 3B, Fig. S1C). GEPIA database was adopted to detect the differential expression and prognostic value of DEGs (Figs. S2A, S3A). In a gene dependent network, the penetrance of a gene represents the number of genes associated with the phenotypic changes it affects. Therefore, genes with higher penetrance are more critical for phenotypic changes. In this work, we selected 16 candidate hub genes (CFTR, CXCL12, IRF4, CD79A, PDGFA, FSTL3, LAMC2, MT2A, PLAUR, SEMA3C, TNFRSF12A, CYP4F12, FUT6, HCG22, HLF, and PTGDS) as the central genes of the network for further analysis because of their high degree of connectivity (P < 0.05).

Screening of hub genes assisted with TCGA database. (A) Hierarchical clustering analysis of the differentially expressed genes between normal and OSCC from TCGA database. (B) GSEA analysis of OSCC in TCGA database. (C) Copy number variation (CNV) frequency of the 16 genes in TCGA cohort. CNV gain, red; CNV loss, blue color. The height of the column represents the alteration frequency. (D) Mutation frequency and classification of the 16 genes in OSCC. (E) Univariate Cox analysis of 16 DEGs with prognostic significance in TCGA OSCC datasets. (F) Multivariate Cox analysis 16 DEGs with prognostic significance in TCGA OSCC datasets.
Copy number variation (CNV) is one of the main factors affecting gene expression abundance in many cancers34,35,36. Thus, we performed CNV frequency analysis for those 16 hub genes in TCGA data. Thus, we analyzed the incidence of CNV and somatic mutations of these hub genes in patients with OSCC in TCGA database, which revealed frequent CNV alterations in these genes, with a higher proportion of copy number losses than gains (Fig. 3C). The CNV mutation frequency of FSTL3 reached 10%, mainly with CNV deletion. This was followed by CFTR, FUT6, HCG22, IRF4, etc., while the CNV mutation frequency in PDGFA, CYP4F12, CXCL12, HLF, LAMC2, PLAUR and CD79A was less than 2.5% (Fig. 3C). In addition, among the 16 hub genes, most of gene mutation were missense mutations. In detail, CFTR accounted for 32.6%, SEMA3C accounted for 16.3%, LAMC2 accounted for 14.0%, CYP4F12 accounted for 11.6 and IRF4 accounted for 9.3% (Fig. 3D).
To further explore whether 16 hub genes were associated with OSCC patient prognosis, we performed univariate and multivariate Cox regression analysis. The results of univariate Cox regression analysis revealed that 13 out of 16 genes were significant risk factors for overall survival (OS) of OSCC patients (P < 0.05) (Fig. 3E). Then, we conducted multivariate Cox regression analysis and selected three genes (P < 0.05) consisting of CD79A, HLF and IRF4 (Fig. 3F). Genes including HLF and IRF4 showed negative coefficients in the multivariate Cox regression analysis, implying low-risk signatures while CD79A showed the opposite effect. Taken together, IRF4 might be used as diagnostic biomarker for transformation of OSF into OSCC.
Identification and validation of hub genes
We calculated the levels of correlation between IRF4 and other 15 hub genes using GEPIA2 dataset and identified significant correlations between IRF4 and 8 out of 15 hub genes. In detail, CD79A, CXCL12, HLF and PTGDS were positively correlated with IRF4, while LAMC2, FSTL3, MT2A and TNFRSF12A were negatively related to IRF4 (Fig. 4A). In order to verify the differential expression of 16 hub genes mRNA, we analyzed the data from the TCGA database and validated that IRF4, CD79A, PDGFA, FSTL3, LAMC2, PLAUR, SEMA3C, TNFRSF12A and MT2A were upregulated in OSCC, while CFTR, CYP4F12, FUT6, HCG22, HLF, and PTGDS were downregulated in OSCC compared with Normal samples (P < 0.01) (Fig. 4B). For the sake of further validating differential expression of hub genes in OSCC and Normal tissues, we extracted total RNA from cell lines (HOK cell line as normal control and HN4 cell line as cancer cell) and fresh tissues (Normal tissues from healthy people and OSCC tissues from OSCC patients) respectively. QRT-PCR of HOK and HN4 showed that the expression of IRF4 in HN4 was almost twice that in HOK (Fig. 4C). QRT-PCR of Normal oral mucosa and OSCC fresh tissues presented a similar result (Fig. 4D). These results were consistent with the previous analysis results, which reminded us that IRF4 might play a positive role in transformation of OSF into OSCC.

Identification and Validation of Hub Genes. (A) Correlation of IRF4 expression with other representative DEGs. (B) Expression data of DEGs in OSCC from TCGA database. (C) The mRNA expressions of DEGs by qRT-PCR in total RNA extracted from Normal and OSCC samples. (D) The mRNA expressions of DEGs by qRT-PCR in total RNA extracted from HOK and HN4 cell lines. ns, No significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Identification of IRF4 as the key immune-related biomarker for transformation of OSF into OSCC through pan cancer analysis and immune infiltration analysis
We further analyzed the expression of IRF4 mRNA in OSCC and Normal tissues using TCGA data sets in GEPIA2, and the data showed higher expression of IRF4 mRNA in OSCC than Normal mucosa (Fig. 5A), which was consistent with previous study. Pan cancer analysis revealed the difference of IRF4 in different Tumor tissues and Normal tissues (Fig. 5B). Compared with Normal tissues, the expression level of IRF4 significantly increased in GBM (glioblastoma multiforme), GBMLGG (glioma), LGG (brain lower grade glioma), ESCA (esophageal carcinoma), STES (stomach and esophageal carcinoma), KIPAN (Pan-kidney cohort), STAD (stomach adenocarcinoma), HNSC (head and neck squamous cell carcinoma), KIRC (kidney renal clear cell carcinoma), SKCM (skin cutaneous melanoma), THCA (thyroid carcinoma), PAAD (pancreatic adenocarcinoma), TGCT (testicular germ cell tumors), ALL (acute lymphoblastic leukemia) and LAML (acute myeloid leukemia), while it was significantly decreased in BRCA (breast invasive carcinoma), CESC (cervical squamous cell carcinoma and endocervical adenocarcinoma), LUAD (lung adenocarcinoma), COAD (colon adenocarcinoma), COADREAD (colon adenocarcinoma/rectum adenocarcinoma), PRAD (prostate adenocarcinoma), LUSC (lung squamous cell carcinoma), LIHC (liver hepatocellular carcinoma), WT (Wilms tumor), BLCA (bladder urothelial carcinoma), READ (rectum adenocarcinoma), OV (ovarian serous cystadenocarcinoma), UCS (uterine carcinosarcoma), ACC (adrenocortical carcinoma) and KICH (kidney chromophobe).

Pan Cancer Analysis and Functional Analysis of IRF4 in OSCC. (A) The mRNA expression of IRF4 in OSCC from TCGA database. (B) The expression status of IRF4 in different cancers. (C) The chord plot showing highly relevant molecules assigned to IRF4. (D) GO enrichment analysis of genes interacting with IRF4. (E) KEGG pathways analysis of genes interacting with IRF4. (F) The correlation of IRF4 and tumor infiltrating immune cells performed via TIMER. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In order to explore possible mechanism of IRF4 in transformation of OSF into OSCC, we constructed IRF4 gene regulatory network including Protein–Protein interaction, Transcription factor-Target gene, miRNA-Target gene and Chemical-Gene interaction with Gendoma (Fig. 5C). GO functional annotation analysis of the proteins interaction with IRF4 indicated that IRF4 related genes mainly regulates immune-related functions including regulation of immune system process, defense response and immune response (Fig. 5D). KEGG pathway analysis suggested that IRF4 related genes were mainly associated with immune cell-related signaling pathway, such as inflammatory bowel disease, Th17 cell differentiation, Th1 and Th2 cell differentiation (Fig. 5E). Meanwhile, GSEA analysis of IRF4 in OSCC also showed a significant correlation immune-related GO terms and KEGG terms (Fig. S4).
To explore the potential immunomodulatory mechanism of IRF4 in the transformation of OSF into OSCC, we used TIMER database to evaluate the correlation between IRF4 expression in OSCC samples and immune infiltrating cells. TIMER data displayed high IRF4 expression was significantly associated with six immune cells (B cells, CD4+ T cells, CD8+ T cells, macrophages, neutrophils, and dendritic cells) in OSCC (Fig. 5F). Tumor purity is an important factor affecting the immune infiltration analysis of clinical tumor samples by genomic methods37,38, TIMER database also indicated that IRF4 expression levels had a significant negative correlation with tumor purity (Fig. 5F). In particular, IRF4 exhibited significantly positive correlation with the two key immune checkpoints CD79A and CD19 (Fig. 6A).

Immune Infiltration Analysis of IRF4 in OSCC. (A) The correlation between IRF4 and representative immune genes performed via TIMER. (B) The relationship between expression, copy number and methylation of IRF4 and immune infiltration levels in pan-cancer from TISIDB. (C) The correlation between IRF4 and multiple immune cells (Pearman’s correlation test). (D) The relevance of different somatic copy number alterations for IRF4 and OSCC infiltration levels from TIMER database. (E) Expression pattern of IRF4 Single-cell RNA in various immune cell types in lymph node from the human protein atlas. (F) The correlation between IRF4 and immune cell type markers from the human protein atlas.
Besides, the relationship between abundance of tumor-infiltrating lymphocytes (TILs) and expression, copy number and methylation of IRF4 in pan-cancer was analyzed through TISIDB. The expression of IRF4 was positively correlated with majority of TIL levels, and the copy number and methylation of IRF4 was negatively related to immune infiltration in OSCC (Fig. 6B). In addition, rho values of spearman correlation test of IRF4 expression and abundance of Activated B cell, Immature B cell, Effector memory CD8 T cell, Type 1 T helper cell, Eosinophil, Myeloid derived suppressor cell and T follicular helper cell were greater than 0.6 (Fig. 6C). At the same time, we found that IRF4 CNV had a closely association with the degree of infiltration of B cell, CD8+ T cell, CD4+ T cell, macrophages, neutrophils and dendritic cell (Fig. 6D). Next, we used the human protein atlas to study the expression pattern of IRF4 Single-cell RNA in immune cell types, and verified the significant correlation between IRF4 and some immune cell types, such as B cells, Dendritic cells, Macrophages, Plasma cells and T cells (Fig. 6E,F).
Validation of IRF4 at translational level
Previous studies have shown a strong correlation between mRNA expression and protein expression, and mRNA upstream regulates protein translation39,40,41. There is a need for consensus on sampling, staining and quantification procedures concerning detection of IRF4 protein expression by IF and IHC staining of tissues from Normal, OSF and OSCC patients to improve reproducibility of studies and confirm bioinformatics results. Here, we detected IRF4 protein expression in different tissues from different patients by IF staining (Fig. 7A,C) and IHC staining (Fig. 7B,D), which showed a consistent, sequential upregulation of IRF4 from Normal buccal mucosa to OSF to OSCC. The results were aligned with previous studies and demonstrated that IRF4 might act as oncogene in transformation of Normal into OSF, Normal into OSCC and OSF into OSCC.

Validation of IRF4 at translational level. (A) IF staining of IRF4 in Normal mucosa, OSF and OSCC tissues. (B) IHC staining of IRF4 in Normal mucosa, OSF and OSCC tissues. (C) The quantitative analysis of IF staining. (D) The quantitative analysis of IHC staining. *p < 0.05; **p < 0.01.
