
Abstract
The adrenal gland responds to heat stress by epinephrine and glucocorticoid release to alleviate the adverse effects. This study investigated the effect of acute heat stress on the protein profile and histone modification in the adrenal gland of layer-type country chickens. A total of 192 roosters were subject to acute heat stress and thereafter classified into a resistant or susceptible group according to body temperature change. The iTRAQ analysis identified 80 differentially expressed proteins, in which the resistant group had a higher level of somatostatin and hydroxy-δ-5-steroid dehydrogenase but a lower parathymosin expression in accordance with the change of serum glucocorticoid levels. Histone modification analysis identified 115 histone markers. The susceptible group had a higher level of tri-methylation of histone H3 lysine 27 (H3K27me3) and showed a positive crosstalk with K36me and K37me in the H3 tails. The differential changes of body temperature projected in physiological regulation at the hypothalamus–pituitary–adrenal axis suggest the genetic heterogeneity in basic metabolic rate and efficiency for heat dissipation to acclimate to thermal stress and maintain body temperature homeostasis. The alteration of adrenal H3K27me3 level was associated with the endocrine function of adrenal gland and may contribute to the thermotolerance of chickens.
Similar content being viewed by others


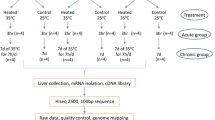
Introduction
Modern chicken breeds dissipate considerable body heat and are sensitive to heat stress due to genetic selection for heightened metabolic activity and meat and egg production1,2. Heat stress leads to adverse alterations of behavioral, physiological, reproductive, and immunological responses, causing significant reduction in feed intake, body weight gain, egg production, and meat and egg quality2,3,4,5,6,7. Diminished growth, disease susceptibility, and high mortality resulting from heat stress account for a large part of the cost of poultry production throughout the world8.
When a behavioral response fails to meet heat loss requirements under a high ambient temperature, the sympathetic–adrenal–medullary axis (SAM axis) and the hypothalamus–pituitary–adrenal axis (HPA axis) are activated to compensate for the thermal imbalance9. Catecholamine (e.g., epinephrine and norepinephrine) and glucocorticoid (GC) release from the SAM axis and HPA axis enhance hepatic glycogenolysis and gluconeogenesis to supply more glucose for energy need in heat stress alleviation10,11.
Proteomics is a powerful tool for improving genetic selection, and has been applied in exploring the biological mechanisms of different tissues in response to heat stress67 for protein expression and histone modification analysis.
Plasma epinephrine and corticosterone analysis
Plasma epinephrine and corticosterone (CORT) levels were measured using Adrenaline Research ELISA (BA E-5100, ImmuSmol SAS, Bordeaux, France) and the Corticosterone ELISA Kit (501,320, Cayman Chemical, Ann Arbor, MI, USA), respectively.
Protein sample preparation, isobaric tags for relative and absolute quantitation (iTRAQ) analysis, and fractionation of peptides
The collected adrenal glands were sliced into small pieces and lysed in O’Farrell’s lysis buffer (9.5 M urea, 65 mM dithiothreitol, 2% v/v Ampholyte 3–10, and 2% NP-40). The samples were sonicated (80 W; four times for 10 s) to dissolute proteins. The homogenates were maintained at 4 °C for 1 h and centrifuged at 14,000 × g at 4 °C for 10 min to obtain supernatants. The supernatants were mixed with 100% trichloroacetic acid (TCA) to obtain a final TCA concentration of 20% and maintained at 4 °C for 1 h with shaking every 15 min. After centrifugation at 14,000 g at 4 °C for 10 min, the precipitated pellets were collected and washed with ice-cold acetone twice. The protein pellets were air-dried for 10 min and dissolved in 4 M urea solution. Protein concentrations were determined using the Bradford method with bovine serum albumin as the standard68.
This study performed iTRAQ labeling according to the manufacturer’s protocol (iTRAQ reagent multiplex kit, Applied Biosystems, Waltham, MA, USA). Five replicated protein samples from the same group were mixed and used for reduction and alkylation, which was followed by overnight digestion with trypsin. The tryptic peptides from the control, resistant, and susceptible groups were labeled with isobaric iTRAQ tags with mass 114, 115, and 116 Da, respectively. The samples were then pooled, dried using a SpeedVac evaporator (Tokyo Rikakikai Co. Ltd., Bunkyo-ku, Tokyo, Japan), and stored at − 80 °C until analysis.
Fractionation of the labeled peptides was performed using an ultraperformance liquid chromatography (UPLC) system (ACQUITY UPLC System, Waters, Milford, MA, USA) and a 2.1 mm × 150 mm × 1.7 µm column with a volume of 0.519 mL (ACQUITY UPLC BEH C18, Waters). The mobile phase was prepared in a gradient with 10 mM ammonium bicarbonate (ABC, pH 10, mobile phase A) and 10 mM ABC/90% acetonitrile (pH 10, mobile phase B). A gradient was created with mobile phase B from 0 to 3% during min 0–5; 3% to 30% during min 5–40; 30% to 70% during min 40–55; and 70% to 0% during min 55–60. The flow rate was 0.2 µL/min. Fractions were collected in 1-min intervals for 1 h duration. Urea solutions in various fractions were removed using C18 ZipTip (Merck, Darmstadt, Germany). All fractions were dried using a SpeedVac evaporator (Tokyo Rikakikai Co. Ltd.) and stored at − 80 °C until analysis.
Protein identification using nano-UPLC–electrospray ionization (ESI)–quadruple time-of-flight (Q-TOF)–MS/MS
A nano-LC–MS/MS system was used to analyze the tryptic peptides. The peptides were separated using an Ultimate 3000 LC RSLC nano-LC system (Dionex-Thermo Scientific, Chelmsford, MA, USA) coupled with a Q-TOF mass spectrometer (ma** column (Acclaim PepMap C18, Dionex-Thermo Scientific) connected to a C18 analyst column (Acclaim PepMap C18, Dionex-Thermo Scientific) for peptide separation. The labeled peptides were eluted using a linear gradient of mobile phase A (2% ACN and 0.1% FA) and mobile phase B (80% ACN and 0.1% FA) applied at a flow rate of 0.3 µL/min for 90 min. The gradient conditions were as follows: 5% to 30% mobile phase B during min 5–65; 30% to 98% mobile phase B during min 65–79, and finally, down to 10% mobile phase B within 1 min.
The mass spectrometer was operated at 50–2000 m/z at 2 Hz, and the 20 most intense ions with 420–2000 m/z in each survey scan were selected for the MS/MS experiment. MS/MS data were acquired from 50 to 2000 m/z at 5–10 Hz. The MS/MS spectra were de novo sequenced and assigned a protein ID by using PEAKS X (Version X, ; Bioinformatics Solutions, Waterloo, Canada) and searched against the NCBInr database (NCBInr 20,180,904 version) for protein identification. The false discovery rate (FDR) of peptide identification was set to be less than 1%. Protein quantification was achieved using PEAKS X with a significant score (− 10logP) > 15 equal to a P-value < 0.03 using FDR-corrected peaks, and at least one unique peptide was detected. Totally, 80 DEPs were identified and quantified using iTRAQ analysis with a 1.3-fold change for a high (> 1.3) or low (< 0.77) level of relative abundance being considered as differentially expressed proteins (DEPs) of the upregulation or downregulation between the two compared groups, respectively. The volcano diagrams and hierarchical clustering of DEPs were generated by PEAKS X software (Bioinformatics Solutions).
Bioinformatics analysis of DEPs
The DEPs among the groups were annotated for their cellular components, biological processes, and molecular functions by using the Gene Ontology database (amigo1.geneontology.org/cgi-bin/amigo/go.cgi).
Western blot analysis
In electrophoresis for protein separation, each well contained a respective sample with 50 µg of proteins. Proteins were transferred onto PVDF (polyvinylidene fluoride) membrane through the wet-transfer method. A mouse anti-HSP70 (clone N27F3-4) monoclonal antibody was purchased from Enzo Life Sciences (New York, USA). A mouse anti-GAPDH (clone 1D4) monoclonal antibody was purchased from Novus Biologicals (Denver, USA). Horseradish peroxidase conjugated secondary antibodies; goat anti-mouse IgG (Beckman Coulter, Brea, CA, USA) was used for to identify the bands reactive to the primary antibodies through an enhanced chemiluminescence reagent (Pierce Biotechnology Inc., Rockford, IL, USA). Primary and secondary antibodies were incubated with membranes at 1:1000 and 1:5000 dilation, respectively. Signaling was quantified by the luminescence image analyzer ImageQuant LAS 4000 (GE Healthcare Life Sciences).
Histone sample preparation, chemical derivatization, trypsin digestion, and desalting
Histones were isolated using a modified protocol69. Briefly, nuclei were isolated with nuclei isolation buffer (NIB; 15 mM Tris, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, and 250 mM sucrose and protease inhibitor cocktail tablet; pH 7.5) and 0.2% NP-40. After they had been cut into small pieces, the adrenal glands in NIB were homogenized using a homogenizer (T 10 basic ULTRA-TURRAX, IKA, Guangzhou, China), which was followed by 10 min incubation on ice. The mixture was centrifuged at 1,000 × g at 4 °C for 10 min, and the resultant nuclei pellets were collected. The pellets were washed with NIB twice. Histones were then acid-extracted from the isolated nuclei by using 0.2 M H2SO4 at 4 °C for 4 h with shaking every 15 min. The histone-containing supernatants were mixed with 100% TCA to a final TCA concentration of 33% and incubated on ice for 1 h. The histone-enriched pellets were washed with ice-cold acetone/0.1% hydrochloric acid and ice-cold acetone and centrifuged to enable pellet collection. The collected pellets were air-dried and reconstituted in double-distilled water. Finally, the histones were purified through centrifugation and quantified for concentration by using the Bradford method with bovine serum albumin as the standard (Peterson, 1983). All samples were dried using a SpeedVac evaporator (Tokyo Rikakikai Co. Ltd.) and dissolved in 40 μL of 50 mM ammonium bicarbonate, which had pH 8 (concentration > 1 μg/μL). Histones were prepared for MS analysis through propionic anhydride chemical derivatization, trypsin digestion, and propionylation of histone peptides at N-termini, as was described by Sidoli et al. (2016). Then, all histone peptides were desalted with C18 ZipTip (Merck), dried using the SpeedVac evaporator, and finally stored at − 80 °C until analysis.
Identification of histone modifications by using nano-UPLC-ESI-Q-TOF–MS/MS
Nano-LC–MS/MS and the protocol for identification of histone modifications were performed as is described in “Acute heat stress modulates adrenal HPTMs” section. Briefly, histone peptides dissolved in 10 µL of loading buffer were separated and eluted using a linear gradient of mobile phase A (2% ACN, 0.1% FA) and mobile phase B (80% ACN, 0.1% FA) applied at a flow rate of 0.3 µL/min for 90 min. The gradient conditions were as follows: 10% to 40% mobile phase B at min 6–74, 40% to 99% mobile phase B at min 74.1–79, and finally, down to 10% mobile phase B within 1 min.
The MS parameters were as described in “Acute heat stress modulates adrenal HPTMs” section. Label-free quantification was performed using the quantitation module of PEAKS X. Modified histone peptides were identified using PEAKS X through the following search parameters: parent mass error tolerance: 80.0 ppm; fragment mass error tolerance: 0.07 Da; enzyme: trypsin; maximum number of missed cleavages: 2; digestion mode: specific; fixed modifications: propionyl (N-term): 56.0; variable modifications: oxidation (M): 15.99, acetylation (K): 42.01, dimethylation (K): 28.03, methylation (K): 14.02, trimethylation (K): 42.05, propionyl (K): 56.03, deamidation (NQ): 0.98, propionylmethyl: 70.04; maximum number of variable PTMs per peptide: 9; reported number of peptides: 5; and data refine dependencies: 1, 4, 3, 2, 5, 6, 7, 8, 9, 10, 11, 12, 14, 13, 15, 16, 17, 19, 18, and 20. The quantification of histone modification was performed using the PEAKS DB database, which provided an overview of all peptides and histone modifications. The relative abundance of a given PTM resulting from single- or co-occurring PTMs was calculated by dividing its intensity by the sum of intensities for all modified and unmodified peptides sharing the same sequence and without missing values. Therefore, the given PTMs could have only a single datum. The quantification of each peptide of co-occurring PTMs on histone H3 was divided by the quantification of all modified and unmodified peptides to obtain a relative quantification of the histone H3 peptide and the crosstalk of PTMs on histone H3.
Statistical analysis
The concentrations of plasma epinephrine and CORT were analyzed using Student’s t test in the Statistical Analysis System (SAS) software70. The normality of the body temperature changes, western blot analysis and relative values of DEPs and HPTMs were assessed using the normality test. Normally distributed data were analyzed using the least squares means procedure, whereas non-normally distributed data were analyzed using the Kruskal–Wallis test.
References
Deeb, N. & Cahanerm, A. Genotype-by-environment interaction with broiler genotypes differing in growth rate. 3. Growth rate and water consumption of broiler progeny from weight-selected versus non selected parents under normal and high ambient temperatures. Poult. Sci. 81, 293–301 (2002).
Pawar, S. S. et al. Assessing and mitigating the impact of heat stress in poultry. Adv. Anim. Vet. Sci. 4, 332–341 (2016).
Lara, L. J. & Rostagno, M. H. Impact of heat stress on poultry production. Animals 3, 356–369 (2013).
Cheng, C. Y. et al. Functional genomics study of acute heat stress response in the small yellow follicles of layer-type chickens. Sci. Rep. 8, 1320 (2018).
Farag, M. R. & Alagawany, M. Physiological alterations of poultry to the high environmental temperature. J. Therm. Biol. 76, 101–106 (2018).
Tu, W. L. et al. Annotation of differential protein expression in the hypothalami of layer-type Taiwan country chickens in response to acute heat stress. J. Therm. Biol. 77, 157–172 (2018).
Wang, S. H. et al. Acute heat stress changes protein expression in the testes of a broiler-type strain of Taiwan country chickens. Anim. Biotechnol. 30, 129–145 (2018).
Mishra, B. & Jha, R. Oxidative stress in the poultry gut: potential challenges and interventions. Front. Vet. Sci. 6, 60 (2019).
Calefi, A. S., Quinteiro-Filho, W. M., Ferreira, A. J. P. & Palermo-Neto, J. Neuroimmunomodulation and heat stress in poultry. Worlds Poult. Sci. J. 73, 493–504 (2017).
Barth, E. et al. Glucose metabolism and catecholamines. Crit. Care. Med. 35, 508–518 (2007).
Opata, A. A., Cheesman, K. C. & Geer, E. B. The Hypothalamic Pituitary Adrenal Axis in Health and Disease: Cushing’s Syndrome and Beyond. Page 3 in Glucocorticoid Regulation of Body Composition and Metabolism (Springer, 2016).
**ng, T. et al. Proteome analysis using isobaric tags for relative and absolute analysis quantitation (iTRAQ) reveals alterations in stress-induced dysfunctional chicken muscle. J. Agric. Food Chem. 65, 2913–2922 (2017).
Ma, D. et al. iTRAQ-based quantitative proteomics analysis of the spleen reveals innate immunity and cell death pathways associated with heat stress in broilers (Gallus gallus). J. Proteom. 196, 11–21 (2019).
Kang, D. R. & Shim, K. S. Proteomic analysis of the protective effect of early heat exposure against chronic heat stress in broilers. Animals 10, 1234 (2020).
Boschetti, E., Hernandez-Castellano, L. E. & Righetti, P. G. Progress in farm animal proteomics: the contribution of combinatorial peptide ligand libraries. J. Proteom. 197, 1–13 (2019).
Kim, G. H., Ryan, J. J., Marsboom, G. & Archer, S. L. Epigenetic mechanisms of pulmonary hypertension. Pulm. Circ. 1, 347–356 (2011).
Ibeagha-Awemu, E. M. & Zhao, X. Epigenetic marks: regulators of livestock phenotypes and conceivable sources of missing variation in livestock improvement programs. Front. Genet. 6, 302 (2015).
Huang, H., Lin, S., Garcia, B. A. & Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 115, 2376–2418 (2015).
Yeh, C. C. Effect of acute heat stress on the blood characteristics of Taiwan country chickens and broilers. J. Chin. Soc. Anim. Sci. 21, 57–66 (1992).
Nawab, A. et al. Heat stress in poultry production: mitigation strategies to overcome the future challenges facing the global poultry industry. J. Therm. Biol. 78, 131–139 (2018).
Sarg, B. et al. Identification of novel post-translational modifications in linker histones from chicken erythrocytes. J. Proteom. 113, 162–177 (2015).
Wu, H., **ao, K. & Tian, Z. Top-down characterization of chicken core histones. J. Proteom. 184, 34–38 (2018).
Kim, T. & Loh, Y. P. Chromogranin A: a surprising link between granule biogenesis and hypertension. J. Clin. Invest. 115, 1711–1713 (2005).
Valdiglesias, V. et al. Is salivary chromogranin A a valid psychological stress biomarker during sensory stimulation in people with advanced dementia?. J. Alzheimers Dis. 55, 1509–1517 (2017).
Escribano, D. et al. Salivary biomarkers to monitor stress due to aggression after weaning in piglets. Res. Vet. Sci. 123, 178–183 (2019).
Pasqua, T. et al. Impact of chromogranin A deficiency on catecholamine storage, catecholamine granule morphology and chromaffin cell energy metabolism in vivo. Cell Tissue Res. 363, 693–712 (2016).
Ericsson, M., Fallahsharoudi, A., Bergquist, J., Kushnir, M. M. & Jensen, P. Domestication effects on behavioural and hormonal responses to acute stress in chickens. Physiol. Behav. 133, 161–169 (2014).
Fallahsharoudi, A. et al. Domestication effects on stress induced steroid secretion and adrenal gene expression in chickens. Sci. Rep. 5, 15345 (2015).
Soleimani, A. F., Zulkifli, I., Omar, A. R. & Raha, A. R. Physiological responses of 3 chicken breeds to acute heat stress. Poult. Sci. 90, 1435–1440 (2011).
Payne, A. H. & Hales, D. B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970 (2004).
Spiga, F., Walker, J. J., Terry, J. R. & Lightman, S. L. HPA axis-rhythms. Comp. Physiol. 4, 1273–1298 (2014).
Hettel, D. & Sharifi, N. HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nat. Rev. Urol. 15, 191–196 (2018).
Artemenko, I. P., Zhao, D., Hales, D. B., Hales, K. H. & Jefcoate, C. R. Mitochondrial processing of newly synthesized steroidogenic acute regulatory protein (StAR), but not total StAR, mediates cholesterol transfer to cytochrome P450 side chain cleavage enzyme in adrenal cells. J. Biol. Chem. 276, 46583–46596 (2001).
Schoneveld, O. J., Gaemers, I. C. & Lamers, W. H. Mechanisms of glucocorticoid signalling. Biochim. Biophys. Acta 1680, 114–128 (2004).
Okamoto, K. & Isohashi, F. Macromolecular translocation inhibitor II (Zn2+-binding protein, parathymosin) interacts with the glucocorticoid receptor and enhances transcription in vivo. J. Biol. Chem. 280, 36986–36993 (2005).
Okamoto, K. et al. A small nuclear acidic protein (MTI-II, Zn2+ binding protein, parathymosin) that inhibits transcriptional activity of NF-κB and its potential application to antiinflammatory drugs. Endocrinology 157, 4973–4986 (2016).
Bram, Z. et al. Does somatostatin have a role in the regulation of cortisol secretion in primary pigmented nodular adrenocortical disease (PPNAD)? A clinical and in vitro investigation. J. Clin. Endocrinol. Metab. 99, 891–901 (2014).
Lin, L. C. & Sibille, E. Somatostatin, neuronal vulnerability and behavioral emotionality. Mol. Psychiatry 20, 377–387 (2015).
Stengel, A. & Tache, Y. F. Activation of brain somatostatin signaling suppresses CRF receptor-mediated stress response. Front. Neurosci. 11, 231 (2017).
Spiegel, R. et al. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur. J. Hum. Genet. 22, 902–906 (2014).
Sheftel, A. D. et al. Humans possess two mitochondrial ferredoxins, FDX1 and FDX2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 11775–11780 (2010).
El-Hattab, A. W. & Scaglia, F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 10, 186–198 (2013).
Maalej, M. et al. Clinical, molecular, and computational analysis in two cases with mitochondrial encephalomyopathy associated with SUCLG1 mutation in a consanguineous family. Biochem. Biophys. Res. Commun. 495, 1730–1737 (2018).
Na, J. et al. Selenophosphate synthetase 1 and its role in redox homeostasis, defense and proliferation. Free Radic. Biol. Med. 127, 190–197 (2018).
Lee, S., Kim, S. M. & Lee, R. T. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid. Redox Signal. 18, 1165–1207 (2013).
Zhang, J., Li, X., Han, X., Liu, R. & Fang, J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 38, 794–808 (2017).
Slimen, I. B. et al. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 30, 513–523 (2014).
Lindquist, S. & Craig, E. A. The heat-shock proteins. Annu. Rev. Genet. 22, 631–677 (1998).
Tomanek, L. Variation in the heat shock response and its implication for predicting the effect of global climate change on species’ biogeographical distribution ranges and metabolic costs. J. Exp. Biol. 213, 971–979 (2010).
Hyland, E. M. et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell Biol. 25, 10060–10070 (2005).
Postberg, J., Forcob, S., Chang, W. J. & Lipps, H. J. The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 10, 259 (2010).
Corso-Diaz, X., Jaeger, C., Chaitankar, V. & Swaroop, A. Epigenetic control of gene regulation during development and disease: a view from the retina. Prog. Retin. Eye Res. 65, 1–27 (2018).
David, S.-A. et al. Genome-wide epigenetic studies in chicken: a review. Epigenomes 1, 20 (2017).
Luo, J. et al. Histone methylation analysis and pathway predictions in chickens after MDV infection. PLoS ONE 7, e41849 (2012).
Mitra, A. et al. Marek’s disease virus infection induces widespread differential chromatin marks in inbred chicken lines. BMC Genom. 13, 557 (2012).
Mitra, A. et al. Histone modifications induced by MDV infection at early cytolytic and latency phases. BMC Genom. 16, 311 (2015).
Sidoli, S., Cheng, L. & Jensen, O. N. Proteomics in chromatin biology and epigenetics: elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteom. 75, 3419–3433 (2012).
Latham, J. A. & Dent, S. Y. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024 (2007).
Chi, P., Allis, C. D. & Wang, G. G. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469 (2010).
Young, N. L., Dimaggio, P. A. & Garcia, B. A. The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67, 3983–4000 (2010).
Schwammle, V. et al. Systems level analysis of histone H3 post-translational modifications (PTMs) reveals features of PTM crosstalk in chromatin regulation. Mol. Cell. Proteom. 15, 2715–2729 (2016).
Knutson, S. K. et al. Synergistic anti-tumor activity of EZH2 inhibitors and glucocorticoid receptor agonists in models of germinal center non-hodgkin lymphomas. PLoS ONE 9, e111840 (2014).
Greulich, F., Hemmer, M. C., Rollins, D. A., Rogatsky, I. & Uhlenhaut, N. H. There goes the neighborhood: assembly of transcriptional complexes during the regulation of metabolism and inflammation by the glucocorticoid receptor. Steroids 114, 7–15 (2016).
Misale, M. S., Janusek, L. W., Tell, D. & Mathews, H. L. Chromatin organization as an indicator of glucocorticoid induced natural killer cell dysfunction. Brain Behav. Immun. 67, 279–289 (2018).
Yang, K. T. et al. Expressed transcripts associated with high rates of egg production in chicken ovarian follicles. Mol. Cell Probes 22, 47–54 (2008).
Zhuang, Z. X., Chen, S. E., Chen, C. F., Lin, E. C. & Huang, S. Y. Genomic regions and pathways associated with thermotolerance in layer-type strain Taiwan indigenous chickens. J. Therm. Biol. 88, 102486 (2020).
Moawad, U. K. & Randa, M. H. Histocytological and histochemical features of the adrenal gland of Adult Egyptian native breeds of chicken (Gallus Gallus domesticus). Beni-Suef Univ. J. Basic Appl. Sci. 6, 199–208 (2017).
Peterson, G. L. Determination of total protein. Methods Enzymol. 91, 95–119 (1983).
Sidoli, S., Bhanu, N. V., Karch, K. R., Wang, X. & Garcia, B. A. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp. 111, e54112 (2016).
SAS Institute Inc. SAS/STAT 9.2 User’s Guide (SAS Institute Inc, 2013).
Acknowledgements
This study was financially supported by the Ministry of Science and Technology (contract nos. MOST105-2321-B-005-014, MOST106-2321-B-005-007 and MOST107-2313-B-005-034), and The iEGG and Animal Biotechnology Center from The Future Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan (contract no. 108-S-0023). The authors also thank Miss Yu-Hui Chen for hel** the western blot analysis.
Author information
Authors and Affiliations
Contributions
H.T.Z., Z.X.Z., C.J.C., C.F.C., S.E.C., S.Y.H. conceived and designed the experiments. H.T.Z., Z.X.Z., H.Y.L., H.L.C., H.C.H. performed the experiments. H.T.Z., C.J.C., H.Y.L. analyzed the data. C.J.C., C.F.C., S.E.C., S.Y.H. contributed reagents/materials/analysis tools. H.T.Z., S.E.C., S.Y.H. wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, HT., Zhuang, ZX., Chen, CJ. et al. Effects of acute heat stress on protein expression and histone modification in the adrenal gland of male layer-type country chickens. Sci Rep 11, 6499 (2021). https://doi.org/10.1038/s41598-021-85868-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85868-1
- Springer Nature Limited