
Abstract
Wheat dwarf bunt is caused by Tilletia controversa Kühn, which is one of the most destructive diseases of wheat worldwide. To explore the interaction of T. controversa and wheat, we analysed the transcriptome profile of spikes of the susceptible wheat cultivar Dongxuan 3, which was subjected to a T. controversa infection and a mock infection. The results obtained from a differential expression analysis of T. controversa-infected plants compared with mock-infected ones showed that 10,867 out of 21,354 genes were upregulated, while 10,487 genes were downregulated, and these genes were enriched in 205 different pathways. Our findings demonstrated that the genes associated with defence against diseases, such as PR-related genes, WRKY transcription factors and mitogen-activated protein kinase genes, were more highly expressed in response to T. controversa infection. Additionally, a number of genes related to physiological attributes were expressed during infection. Three pathways were differentiated based on the characteristics of gene ontology classification. KEGG enrichment analysis showed that twenty genes were expressed differentially during the infection of wheat with T. controversa. Notable changes were observed in the transcriptomes of wheat plants after infection. The results of this study may help to elucidate the mechanism governing the interactions between this pathogen and wheat plants and may facilitate the development of new methods to increase the resistance level of wheat against T. controversa, including the overexpression of defence-related genes.
Similar content being viewed by others

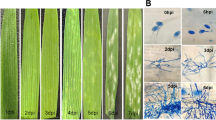

Introduction
Wheat (Triticum aestivum L.) is an important staple food crop in most agricultural regions1,2. Wheat crops are affected by many fungal diseases, among which dwarf bunt of wheat (DBW) caused by Tilletia controversa Kühn is considered to be very dangerous in the majority of wheat-cultivating regions worldwide3,4. In the past decade, DBW has devastated several hundreds of acres (1 hectare = 2.471 acres) of wheat in cold areas of the world5,6,7,8,9. A typical symptom of DBW infection is the replacement of grains with bunt sori, which contain millions of dark black teliospores. The infected grains have reduced quality and market ability, due to the characteristic odour of trimethylamine10,11. Also, the minced flour from DBW-infected grains has a bad taste and fishy smell12,13,14. Because of the quarantine importance of DBW in many countries worldwide regarding wheat production15,16, most studies have focused on pathogen detection17,18,19,20,21. Assessments of the risk of establishing the disease in China was also reported22,23, and these studies mentioned that there were several very high-risk zones in the main winter wheat growing regions, including the northern **njiang Uygur Autonomous Region, Tibet, Sichuan, Henan, Shandong, and the Huaihe River Valley. However, few studies have investigated the molecular mechanism governing the interactions of wheat with T. controversa. The primary control measure taken against DBW was the use of disease-free grains for sowing and the application of fungicides16. Thus, breeding wheat varieties with durable resistance to DBW is one of the most environmentally friendly approaches and may be the most beneficial and effective strategy for disease management, but the development of resistant wheat varieties is laborious, difficult, and time-consuming. Several preliminary genetic studies have been performed, but they have resulted in the discovery of only a small number of genes contributing to control of DBW in field conditions5.
Characterizing the response of wheat to T. controversa infection is important for determining the mechanisms of wheat resistance to T. controversa infection and develo** suitable approaches for DBW control. Multilayering occurs between pathogens and plants, and considerable research has been conducted to elucidate the molecular mechanism underlying the interactions between pathogens and plants24,25. With advances in plant science, plants have been determined to have many different novel strategies to defend themselves against various levels of invaders, including bacteria, viruses and different classes of fungi26,27. These novel strategies of plants contain more complex and linked mechanisms, which increase the correlation of plant pathogen interactions, namely, effector triggered immunity (ETI) and PAMP-triggered immunity (PTI)28,29,30,31,32,33, and some important plant hormones, including salicylic acid, jasmonic acid and ethylene34,35,36,37. PTI is the initial response of plants under pathogen attack when host receptors diagnose the pathogen-derived pathogen-associated molecular pattern (PAMP), whereas ETI is triggered by the interface between a “resistance” protein and a pathogenic effector38,39,40. Meanwhile, the response to fungal infection of wheat is very complex, and many morpho-physiological processes are involved, but system-level transcriptomic studies may enhance our basic understanding of the response of the host to pathogens. High-throughput gene expression analysis using the most advanced molecular biological technique (RNA-Seq) represents a very powerful tool for transcriptomic characterization of plants during the interaction of pathogens and plants41,42. RNA-Seq has been performed successfully in many other crops and pathogens, including tomato and Xanthomonas perforans race T343, the chestnut and Cryphonectria parasitica strain EP15544, potato and Phytophthora infestants45, banana and Fusarium oxysporum f. sp. cubense46, peach and Xanthomonas arboricola pv. pruni47, soybean and Fusarium oxysporum48 and mango and Fusarium mangiferae49. Previous studies demonstrated that several genes were involved in defence mechanisms and resistance-associated signal transduction during plant pathogen interactions50. In this study, RNA-Seq was performed to analyse the changes in gene expression and signal transduction in response to T. controversa infection. Differentially expressed genes (DEGs) involved in resistance to DBW were investigated after successful infection with T. controversa. This approach has led to a greater understanding of the cellular and complex molecular events associated with DBW and provided a basis for further studies on biotechnology and breeding for resistance to DBW disease51,52. Many studies have been performed in susceptible cultivars of various crops to examine gene expression over a time course after pathogen infection53. DBW is a quarantine significant disease that causes severe losses under optimum conditions in wheat. However, very little is known regarding the molecular and cellular mechanisms underlying the interactions between wheat and T. controversa. This study was performed to elucidate the interactions between wheat and T. controversa to develop effective strategies for controlling important wheat diseases. To the best of our knowledge, this study is the first to determine the transcriptome changes in wheat after T. controversa infection.
Materials and methods
Fungal materials and culture
The T. controversa isolate was provided by Blair Goates, the United States Department of Agriculture (USDA), Agricultural Research Service (ARS), Aberdeen, Idaho, USA. We purified the isolate using a single isolation method and identified the isolate as race 2, which was highly aggressive. The cultivation methods for the hyphae of T. controversa used for inoculation were prepared according a previously described protocol54.
Inoculation of wheat plants with T. controversa
Dongxuan 3, a winter wheat cultivar that is highly susceptible to T. controversa, was used in this study. Seeds were surface-sterilized with 30% NaClO for 1 min, washed with sterile water 3 times and kept in plates with moist filter paper at 5 °C for one month to vernalize. After vernalization, seedlings were transplanted into pots filled with organic matter and soil at a ratio of 1:2% and were grown in growth chambers (Percival, ARC-36VL-LT, USA). Wheat seedlings were grown in a 14 h light/10 h dark cycle at 5 °C at the tillering stage and at 25 °C at the boot stage. At the early boot stage, when the young tassels of wheat were still wrapped by leaf sheaths, the spikes were injected with 1 ml inoculum suspensions of T. controversa. The suspensions contained infectious hyphae at a concentration of 106 cfu/ml and had an OD600 of 0.15. Inoculation was repeated 3 times after a one-day interval. For the mock infection, plants injected with sterilized ddH2O were grown under the same conditions. The samples (with spikes measuring 6.0 ± 0.5 cm in length) were collected from both T. controversa-infected and mock-infected plants (Supplementary Fig. 1), with three biological replicates being employed for each treatment. Six samples were collected and stored immediately at -80 °C for further use.
Extraction and purification of RNA
Total RNA was extracted based on the manufacturer’s protocol of the mirVana miRNA Isolation Kit (Ambion, TX, USA). RNA concentration was measured with a Qubit RNA Assay Kit in a Qubit 3.0 Fluorometer (Life Technology, CA, USA). The samples that exhibited an A260/A280 of 1.8 to 2.1 and an A260/A230 > 2.0 were chosen for further analysis. Furthermore, the integrity of each sample was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Library preparation for RNA-Seq and sequencing
Total RNA (1 µg) of each sample of mock- and T. controversa-infected plants was analysed for library construction. The mRNA was purified by using oligo (dT) magnetic beads, and sequencing libraries were generated with the TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA) by following all instructions mentioned. The mRNAs were crushed into very small fragments under high temperature in the presence of a fragmentation buffer solution. First-strand complementary DNA was obtained using a solution of random oligonucleotides and SuperScript II reverse transcriptase (Illumina, San Diego, CA, USA); similarly, second-strand complementary DNA was obtained using RNase H and DNA polymerase. A QIAquick PCR kit was employed for purification of cDNA fragments. Then, these cDNA fragments were washed with EB buffer for the addition of end-repair poly (A) and ligated with special sequencing adapters. The final cDNA library was constructed by purification of the cDNA small fragments, which were enriched by PCR products.
Library examination and sequencing
The constructed cDNA library was validated by using the Qubit RNA Assay Kit in Qubit 3.0 for initial quantification. The insert size was determined using a Bioanalyzer 2100 Agilent system (Agilent, Santa Clara, CA, USA). Furthermore, the insert was amplified using qPCR (7500, ABI, USA). The clustering of every sample was performed on Generation systems (Illumina, USA) following a previously described protocol. The prepared library was loaded onto an Illumina HiSeq X Ten platform with 150-bp paired-end technology.
Quality control and map**
The raw data of this experiment were further processed using Trimmomatic (trimmer for Illumina sequence data, Version 0.32)55. The reads containing adapter sequences and reads with low quality (those in which more than 50% of bases presented quality of ≤ 10) and poly-N (unrecognized bases) were removed to obtain clean reads. Every downstream analysis was performed based on clear data with significantly high quality. The clean reads were mapped to the reference genome (https://www.ebi.ac.uk/ena/data/view/GCA_900519105.1) using hisat256 with the parameters set by the system.
Gene-level quantification and identification of differentially expressed genes (DEGs)
The FPKM value of every gene was analysed and calculated by using cufflinks (version 2.2.1)57,58, and every read count of all genes was obtained by HTSeq-count59. Additionally, the DEGs of this study were recognized by using the DESeq60 technique. Furthermore, the FDR ˂ 0.05, and at least a two-fold change (> 1 or < − 1 in log2 ratio value) was set as the threshold for DEGs. Hierarchical cluster analysis (HCA) of all DEGs was performed to explore gene expression patterns.
Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) enrichment analysis
KEGG (https://www.kegg.jp/kegg/kegg1.html) pathway analysis was performed by using GPSeq, and GO enrichment analysis was performed, with FDR < 0.05 representing the significantly expressed genes61,62.
Validation of RNA-Seq results by quantitative real-time PCR analysis
Eight transcripts with various expression levels demonstrated by RNA sequencing were randomly selected for proof by qRT-PCR. Total RNA was extracted from three T. controversa-infected spikes and three mock-infected spikes. First-strand cDNA was synthesized by using 2000 ng µl−1 purified total RNA, RT/RI enzyme and oligo (dT)18 primer (TransGen Biotech, Bei**g, China) following the instructions of the manufacturer. The primers employed in this experiment are listed in Table S1. Actin was used as an internal control in this experiment. RT-qPCR was performed using Top Green qPCR SuperMix (TransGen, China) in a volume of 20 µL by following the instructions of the manufacturer and applied to the QuantStudio 5 instrument, which was part of a real-time PCR system (Applied Biosystems, Bei**g, China). Three technical replicates were employed for every gene. All genes were run on a 96-well QuantStudio 5 (Applied Biosystems, Bei**g, China) with the following conditions: pre-denaturation at 95 °C for 10 min and 40 cycles (95 °C for 15 s, 58 °C for 30 s and 72 °C for 30 s). The 2−ΔΔCt method63 was employed to calculate the expression level of every gene.
Results
Confirmation of T. controversa infection in wheat plants
After inoculation with T. controversa, symptoms appeared in the spike with the formation of black teliospores. Specifically, the heads of infected plants were thicker and wider and squarrose. The florets were filled with bunt balls (sori). These bunt balls replaced the grains and produced black teliospores with a rotten fish-like odour. To identify the genes with changed expression following infection by T. controversa and to detect any change in gene expression levels after infection, RNA extraction was performed to investigate the changes in wheat at the transcriptomic level.
Transcriptomic analysis of RNA-Seq data
Based on RNA-Seq, we identified alterations in wheat genes when the spike was infected by T. controversa. Six cDNA libraries (three T. controversa-infected and three mock-infected) were sequenced. Raw reads were trimmed by removing empty reads, adaptor sequences and low-quality sequences. Approximately 55.95, 50.30 and 52.45 million raw reads were obtained from the mock-infected plants CK-1, CK-2 and CK-3, respectively, and raw reads in T. controversa-infected plants were 58.54, 58.50 and 53.72 million in T. controversa (Inoculated-1), T. controversa (Inoculated-2) and T. controversa (Inoculated-3), respectively. Similarly, 54.55, 49.23 and 51.11 million clean reads (high-quality reads) were obtained from mock-infected plants, and 55.72, 55.55 and 51.04 million reads were obtained from Inoculated-1, Inoculated-2 and Inoculated-3, respectively. Multiple and unique maps of the abovementioned transcripts ranged from 7.05 to 11.80% and 79.56 to 87.05%, respectively. Additionally, the Q30 and GC of these transcripts ranged from 94.15 to 96.02% and 50.84 to 55.66%, respectively (Table 1). Next, the differentially expressed genes were recognized by comparing the FPKM value of every gene between CK and T. controversa (Inoculated) samples. For CK-1 DEGs, 9496 (FPKM ˃ = 10), 33,690 (FPKM 1–10), 6827 (FPKM 0.5–1), and 57,532 (FPKM 0.–0.5) genes were differentially expressed. An approximately similar response was observed in CK-2 and CK-3 samples. For Inoculated-1 DEGs, 5150 (FPKM ˃ = 10), 27,077 (FPKM 1–10), 8799 (FPKM 0.5–1), and 66,499 (FPKM 0.-0.5) genes were differentially expressed. The differential expression of genes in CK and T. controversa indicates the genetic difference between mock- and T. controversa-infected plants (Fig. 1). Additionally, three biological replicates of every sample were clustered together. Sample-to-sample clustering analysis demonstrated that the gene expression level between replicates was reproducible and that batch effects were controlled (Fig. 2). Furthermore, principal component analysis (PCA) was performed for both mock- and T. controversa-infected samples. The mock was located around the junction of the second and third quadrants, and the T. controversa infection was located around the junction of the first and fourth quadrants, indicating that there is good reproducibility among the biological replicates of the same treatments but differences between the treatments (Fig. 3).

Summary of differentially expressed genes (DEGs). Numbers of DEGs between CK and T. controversa infection. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.

Sample-to-sample clustering analysis for checking batch effects and their similarity. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.

Principal component analysis (PCA) for gene expression patterns. The first and second PCAs explained 99.11% and 0.47% of the variance, respectively. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.
Identification of differentially expressed genes (DEGs)
The differentially expressed genes were recognized in T. controversa-infected and mock-infected libraries. In this comparison, 10,867 (up-regulated) and 10,487 (down-regulated) genes were expressed (Fig. 4; Table S2). To elucidate the transcriptional changes occurring after T. controversa infection, we demonstrated the expression pattern by using hierarchical clustering analysis. On behalf of the analysis, the expression levels of T. controversa-infected and mock-infected plants were different from each other but were similar in the replication of T. controversa-infected and mock-infected plants. There were more up-regulated genes than down-regulated genes in T. controversa-infected plants (Fig. 5).

Significant differentially expressed genes (DEGs) in T. controversa-infected vs. mock libraries. Up- or downregulated DEGs in response to T. controversa infection. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.

Hierarchical clustering heatmap of DEGs according to changes in expression in response to T. controversa infection. Each column shows a library, and each row shows DEG expression. The colours blue, white and red indicate low, medium, and high expression patterns of genes, respectively. CK means the mock plants, inoculated mean the plants were infected by T. controversa.
Gene ontology (GO) enrichment analysis of DEGs
GO enrichment analysis of DEGs can demonstrate the function of genes. GO was categorized into three main domains based on their functions: biological process, cellular component and molecular function. Our results showed that in the biological process category, during the comparison of T. controversa-infected and mock-infected plants, GO was mainly associated with cellular process, metabolic process, multicellular organismal process, regulation of biological process, reproduction, reproduction process, response to stimulus, and single-organism process. Meanwhile, in the cellular component category, DEGs were primarily associated with cell, cell part, extracellular region, macromolecular complex, membrane, membrane part, organelle, and organelle part. In the molecular function category, DEGs primarily mapped with binding, catalytic activity, enzyme regulator activity, nucleic acid binding transcription factor activity, structural membrane activity and transporter activity (Fig. 6). In contrast, biological adhesion, cell killing and locomotion were observed to be uniquely enriched in biological process, while extracellular matrix, extracellular matrix part and nucleoid were uniquely enriched in cellular component and metallochaperone activity, protein tag, receptor regulator activity and translation regulator activity were determined to be uniquely enriched in molecular function (Fig. 6).

Gene ontology (GO) enrichment analysis of significant DEGs of T. controversa-infected and mock libraries. Annotations are grouped by biological process, cellular component, and molecular function. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.
KEGG enrichment analysis of DEGs
KEGG analyses were performed to better understand the molecular associations among the DEGs. For DEGs between T. controversa-infected and mock-infected plants, 205 different pathways were identified (Table S3). However, the top 20 KEGG enrichment pathways of peroxisomes, FoxO signalling pathway, DNA replication, biosynthesis of amino acids, carbon metabolism, carbon fixation in photosynthetic organisms, methane metabolism, glyoxylate and dicarboxylate metabolism, chloroalkane and chloroalkene degradation, pyruvate metabolism, starch and sucrose metabolism, lysine degradation, valine, leucine and isoleucine degradation, glycine-serine-and threonine metabolism, photosynthesis-antenna proteins, cutin, suberine and wax biosynthesis, fatty acid degradation and glycolysis/gluconeogenesis were primarily activated. The pathway of biosynthesis of siderophore group nonribosomal peptides was activated slightly during the interaction (Fig. 7).

KEGG enrichment analysis scatter plot representing pathways of DEGs in response to T. controversa infection. The colours blue, white and red indicate low, medium, and high expression patterns of genes, respectively. CK indicates mock plants, and Inoculated indicates plants infected by T. controversa.
Differential expression of pathogenesis-related genes after T. controversa infection
Previous studies have shown that PR genes encoding 1,3-glucanases, chitinases and thaumatin-like proteins enhanced the resistance of wheat against various fungal pathogens64. By comparing the transcriptome results obtained in T. controversa-infected plants with those of mock-infected cells, it was found that the transcriptional level of PR genes changed. The results showed that 7 pathogenesis-related genes, 23 thaumatin-like genes, 28 chitinase genes, 121 peroxidase genes, and 36 glucanase genes changed during T. controversa infection. Most of these genes were up-regulated (Table S4).
Differential expression of WRKY transcription factors after T. controversa infection
Following T. controversa infection, we identified significant DEGs of WRKY transcription factors in T. controversa-infected libraries compared with mock-infected ones. Most WRKY transcription factors showed up-regulation after T. controversa infection. The results showed that 44 out of 57 and 13 out of 57 WRKY transcription factors were up-regulated and down-regulated, respectively (Table S5).
Differential expression of protein kinase genes after T. controversa infection
Following T. controversa infection, we identified significant DEGs of protein kinase genes in T. controversa-infected libraries compared to mock-infected libraries. The results showed that 29 calcium-dependent protein kinases, 35 CBL-interacting protein kinases, 7 cold-responsive protein kinases, 11 cyclin kinases, 58 cysteine-rich receptor-like protein kinases, 48 G-type lectin S-receptor-like serine/threonine-protein kinases, 26 leucine-rich repeat receptor-like protein kinases, 21 LRR receptor-like serine/threonine-protein kinases, 50 mitogen-activated protein kinases, 7 probable inactive leucine-rich repeat receptors, 21 probable leucine-rich repeat receptor-like protein kinases, 55 probable LRR receptor-like serine/threonine-protein kinases, 22 probable receptor-like protein kinases, 66 probable serine/threonine-protein kinases, 10 proline-rich receptor-like protein kinases, 8 putative cysteine-rich receptor-like protein kinases, 9 receptor protein kinases, 143 serine/threonine-protein kinases, 10 shaggy-related protein kinases, 15 SNF1-related protein kinases and 110 changed after infection (Table S6).
Quantitative real-time PCR
To verify the changes in expression level exhibited by the identified DEGs in response to T. controversa infection, the expression levels of eight genes examined by quantitative real-time PCR (qRT-PCR). The expression pattern of validated genes was similar to the results obtained from RNA-Seq (Table 2). The qRT-PCR results showed that seven genes were up-regulated and Lipase was determined to be down-regulated by both RNA-Seq and qRT-PCR analyses. Hence, the qRT-PCR results confirmed the RNA-Seq data.
Discussion
In the earliest studies of T. controversa and wheat, microscopic studies were performed to observe the structural changes in wheat after infection. In this research, we examined the plant defence responses in wheat following infection by T. controversa. We investigated interactions between susceptible wheat cultivars (Dongxuan 3) and T. controversa. We hypothesized that in these interactions, wheat transcriptomic changes are associated with the plant response to infection; thus, diseases appeared. Therefore, in this experiment, we employed RNA-Seq to perform a transcriptomic study of wheat following T. controversa infection and analysed the changes in the expression levels of genes in mock- and T. controversa-infected wheat plants. Our results demonstrated significantly differentially expressed genes between mock- and T. controversa-infected libraries.
Pathogenesis-related (PR) genes play a key role in the defence mechanisms of plants against biotic factors64,65,66,67. Overexpression of the PR genes encrypting pathogenesis-related proteins, thaumatin-like proteins, chitinases, peroxidases and glucanases increases resistance to various pathogens in different crops11,68,69. We compared the transcription level between pathogen-infected and mock-infected plants at the flowering stage. The transcription levels of 215 PR genes were changed by T. controversa infection (Table S4), including seven pathogenesis-related proteins, twenty-three thaumatin-like proteins, twenty-eight chitinase proteins, one hundred twenty-one peroxidase proteins and thirty-six glucanase proteins. Most of the one hundred thirty-five genes were up-regulated, and eighty were down-regulated. Together, these defence-related proteins might play a role in disease suppression against T. controversa.
WRKY transcription factors represent the largest protein family in plants, can activate different defence mechanisms and play pivotal roles in regulating defence genes35,70,71,72,73,74. DEG analysis of T. controversa-infected libraries versus mock-infected libraries showed that fifty-seven WRKY genes were expressed. Forty-four WRKY transcription factors were up-regulated, while thirteen were down-regulated. Some of the up-regulated WRKY transcription factors, including WRKY transcription factors 23 and 27, play roles in the resistance to biotic diseases (Table S5)75,76,77,78.
Pattern recognition receptors (PRRs) have been developed to recognize MAMPs/PAMPs (microbe/pathogen-associated molecular patterns), which are conserved small molecules present across a broad classification of microbes. These plant receptors belong to the receptor-like kinase (RLK) family79,80,81. In our study, 761 different protein kinases were expressed in wheat ears after T. controversa infection (Table S6). Most kinase proteins were down-regulated after T. controversa infection. Interestingly, cysteine-rich receptor-like protein kinase, leucine-rich repeat receptor protein kinase, probable inactive leucine-rich repeat receptor, probable LRR receptor-like serine/threonine-protein kinase, probable receptor-like protein kinase and proline-rich receptor-like protein kinase were almost completely down-regulated after infection. These consequences indicate that T. controversa releases and carries effectors into wheat ear cells during attack to overcome the immune signalling pattern of plants, thereby leading to DBW. Our results concerning kinase proteins were similar to those obtained by Hosseini82 in their studies of another pathogen. Mitogen-activated protein kinase (MAPK) genes have been investigated in the plant response to fungal pathogens83. In our study, we found that fifty MAPK genes were expressed after T. controversa infection (Table S6), which suggests that MAPK genes play a role in wheat resistance to T. controversa infection.
According to GO enrichment analysis of differentially expressed genes in mock-infected and T. controversa-infected libraries, GO was categorized into three main domains based on their functions: biological process, cellular component, and molecular function. (Fig. 6). The results showed that cellular process and metabolic process had the highest number of DEGs during plant pathogen interactions. The GO results support the hypothesis that plant pathogens cause changes in the primary (plant growth and development) and secondary (induction of defence programme) metabolisms of the plants. Once plants are infected by pathogens, plant metabolism tends to expend more energy on plant defence activation compared with growth, development, cellular maintenance and reproduction84,85,86,87. This phenomenon suggests that the DEG metabolic process is related to the pathogenic mechanism of T. controversa, as well as plant-pathogen interactions.
KEGG pathway analysis demonstrated that most DEGs were characterized by carbon metabolism, starch and sucrose metabolism, biosynthesis of amino acids and glycolysis/gluconeogenesis pathways (Fig. 7; Table S3). Biosynthesis of the siderophore group nonribosomal peptide pathway included down-regulated genes that encode ferric iron acquisition in many microorganisms. Iron plays a role in microbial proliferation and growth. Thus, iron are supposed to play a key role in disease development88,89. Microbial ferric iron reductase is a key enzyme that degrades ferric iron in microbes90. Our results are in kee** with the down-regulation of microbial ferric iron reductase due to fungal infection91,92. The results obtained from KEGG analysis of DEGs showed that significant DEGs were annotated to 205 different pathways, which suggests that T. controversa infection affects various biological functions of wheat (Table S3). Dwarfing is an important symptom of DBW; changes in gene expression related to morpho-physiological characteristics, especially plant height, are notable in wheat infected by T. controversa and might be involved in dwarfing symptoms. Our results showed that the expression of cytochrome P450 changed due to T. controversa infection (Table S3). Cytochrome P450 has various biosynthetic activities and plays a positive role in plant growth and development by producing gibberellins and brassinosteroids93,94.
Overall, our findings provide a genome-wide gene expression profile for wheat plants infected with T. controversa and may help to elucidate the regulatory mechanisms governing the response of wheat to this pathogen.
References
Campos, P. et al. Phosphorus acquisition efficiency related to root traits: Is mycorrhizal symbiosis a key factor to wheat and barley crop**?. Front. Plant Sci. 9, 752 (2018).
Patil, S., Brennan, M., Mason, S. & Brennan, C. The effects of fortification of legumes and extrusion on the protein digestibility of wheat based snack. Foods 5, 26 (2016).
Pieczul, K., Perek, A. & Kubiak, K. Detection of Tilletia caries, Tilletia laevis and Tilletia controversa wheat grain contamination using loop-mediated isothermal DNA amplification (LAMP). J. Microbiol. Methods 154, 141–146 (2018).
Liu, J., Li, C., Muhae-ud-din, G., Liu, T. & Chen, W. Development of the droplet digital PCR to detect the teliospores of Tilletia controversa Kühn in the soil with greatly enhanced sensitivity. Front. Micro. 11, 4 (2020).
Goates, B. J. Identification of new pathogenic races of common bunt and dwarf bunt fungi, and evaluation of known races using an expanded set of differential wheat lines. Plant Dis. 96, 361–369 (2012).
Toh, S. S. & Perlin, M. H. Resurgence of less-studied smut fungi as models of phytopathogenesis in the omics age. Phytopathol. 106, 1244–1254 (2016).
Bokore, F. E. et al. Map** quantitative trait loci associated with common bunt resistance in a spring wheat (Triticum aestivum L.) variety Lillian. Theor. Appl. Genet. 132, 3023–3033 (2019). https://doi.org/10.1007/s00122-019-03403-3.
Laroche, A. et al. Development of a PCR marker for rapid identification of the Bt-10 gene for common bunt resistance in wheat. Genome 43, 217–223 (2000).
Wilcoxson, R. D and Saari, E.E. Bunt and smut diseases of wheat: concepts and methods of disease management. CMMYT 4, 12–25 (1996)
Borgen, A. Organic seed treatment to control common bunt (Tilletia tritici) in wheat. Seed Test. Int. 128, 8–9 (2004).
Gaudet, D. A., Lu, Z. X., Leggett, F., Puchalski, B. & Laroche, A. Compatible and incompatible interactions in wheat involving the Bt-10 gene for resistance to Tilletia tritici, the common bunt pathogen. Phytopathol. 97, 1397–1405 (2007).
Kochanová, M., Zouhar, M., Prokinová, E. & Ryšánek, P. Detection of Tilletia controversa and Tilletia caries in wheat by PCR method. Plant Soil Environ. 50, 75–77 (2004).
Chen, J. et al. A novel QTL associated with dwarf bunt resistance in Idaho 444 winter wheat. Theor. Appl. Genet. 129, 2313–2322 (2016).
Nguyen, H. D. T., Sultana, T., Kesanakurti, P. & Hambleton, S. Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus. 10, 4 (2019).
Hoffmann, J. A. Bunt of wheat. Plant Dis. 66, 979–986 (1982).
Mathre, D. E. Dwarf bunt: politics, identification, and biology. Annu. Rev. Phytopathol. 34, 67–85 (1996).
Gao, L. et al. Development of a SCAR marker for molecular detection and diagnosis of Tilletia controversa Kühn, the causal fungus of wheat dwarf bunt. World J. Microbiol. Biotechnol. 30, 3185–3195 (2014).
Liu, J. H., Gao, L., Liu, T. G. & Chen, W. Q. Development of a sequence-characterized amplified region marker for diagnosis of dwarf bunt of wheat and detection of Tilletia controversa Kühn. Lett. Appl. Microbiol. 49, 235–240 (2009).
Gao, L., Chen, W. Q. & Liu, T. G. Development of a SCAR marker by inter-simple sequence repeat for diagnosis of dwarf bunt of wheat and detection of Tilletia controversa Kühn. Folia Microbiol. 55, 258–264 (2010).
Gao, L., Chen, W. & Liu, T. An ISSR-based approach for the molecular detection and diagnosis of dwarf bunt of wheat, caused by Tilletia controversa Kühn. J. Phytopathol. 159, 155–158 (2011).
Li, C. et al. iTRAQ-based proteomic analysis of wheat bunt fungi Tilletia controversa, T. caries, and T. foetida. Curr. Microbiol. 75, 1103–1107 (2018).
Jia, W. M. et al. Assessment of risk of establishment of whaet dwarf bunt (Tilletia controversa) in China. J. Integr. Agric. 12, 87–94 (2013).
Wei, S., Zhang, Z. & Zhang, Y. Evaluation on the establishment potential of wheat dwarf bunt with bioclimatic analogical distance model. Acta Agric. Universitatis Pekinensis. 21, 127 (1995) ((In Chinese)).
Boyd, L. A., Ridout, C., O’Sullivan, D. M., Leach, J. E. & Leung, H. Plant-pathogen interactions: disease resistance in modern agriculture. Trends Genet. 29, 233–240 (2013).
Orłowska, E., Llorente, B. & Cvitanich, C. Plant integrity: an important factor in plant-pathogen interactions. Plant Signal. Behav. 8, e22513 (2013).
Dangl, J. L. & Jones, J. D. G. Plant pathogens and integrated defence responses to infection. Nature 411, 826–833 (2001).
Wise, R. P., Moscou, M. J., Bogdanove, A. J. & Whitham, S. A. Transcript profiling in host–pathogen interactions. Annu. Rev. Phytopathol. 45, 329–369 (2007).
Bonas, U. & Lahaye, T. Plant disease resistance triggered by pathogen-derived molecules: Refined models of specific recognition. Curr. Opin. Microbiol. 5, 44–50 (2002).
Göhre, V. & Robatzek, S. Breaking the barriers: microbial effector molecules subvert plant immunity. Annu. Rev. Phytopathol. 46, 189–215 (2008).
Bigeard, J., Colcombet, J. & Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 8, 521–539 (2015).
Zhang, J. et al. Effector-triggered and pathogen-associated molecular pattern – triggered immunity differentially contribute to basal resistance to Pseudomonas syringae. Mol. Plant Microbe Interact. 23, 940–948 (2010).
Sun, K. et al. Silencing of six susceptibility genes results in potato late blight resistance. Transgenic Res. 25, 731–742. https://doi.org/10.1007/s11248-016-9964-2 (2016).
Thomma, B. P. H. J., Nu, T. & Joosten, M. H. A. J. Of pamps and effectors: the blurred PTI-ETI dichotomy. Plant Cell 23, 4–15 (2011).
Denancé, N. Disease resistance or growth: the role of plant hormones in balancing immune responses and fitness costs. Front. Plant Sci. 4, 155 (2013).
Sato, M. et al. Network modeling reveals prevalent negative regulatory relationships between signaling sectors in Arabidopsis immune signaling. PLoS Pathog. 6, e1001011 (2010).
Bari, R. & Jones, J. D. G. Role of plant hormones in plant defence responses. Plant Mol. Biol. 69, 473–488 (2009).
Alazem, M. & Lin, N. S. Roles of plant hormones in the regulation of host-virus interactions. Mol. Plant Pathol. 16, 529–540 (2015).
Sarris, P. F. et al. Phytobacterial type 111 effectors hopx1, hopab1 and hopf2 enhance sense-post-transcriptional gene silencing independently of plant r gene-effector recognition. Mol. Plant Microbe Interact. 24, 907–917 (2011).
Thines, M. An evolutionary framework for host shifts—jum** ships for survival. New Phytol. 224, 605–617 (2019).
Kretschmer, M., Damoo, D., Djamei, A. & Kronstad, J. Chloroplasts and plant immunity: where are the fungal effectors?. Pathogens 9, 19 (2020).
Wang et al., M. G. & M. S. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Massart, S., Olmos, A., Jijakli, H. & Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 188, 90–96 (2014).
Du, H., Wang, Y., Yang, J. & Yang, W. Comparative transcriptome analysis of resistant and susceptible tomato lines in response to infection by Xanthomonas perforans race T3. Front. Plant Sci. 6, 1173 (2015).
Barakat, A. et al. Chestnut resistance to the blight disease : insights from transcriptome analysis. BMC Plant Biol. 12, 38 (2012).
Gao, L., Tu, Z. J., Millett, B. P. & Bradeen, J. M. Insights into organ-specific pathogen defense responses in plants: RNA-seq analysis of potato tuber- Phytophthora infestans interactions. BMC Genomics 14, 340 (2013).
Li, C. et al. Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genomics 14, 851 (2013).
Pothier, F. et al. Comparative RNA-seq analysis of early-infected peach leaves by the invasive phytopathogen Xanthomonas arboricola pv. pruni. PloS One 8, e54196 (2013).
Lanubile, A., Muppirala, U. K., Severin, A. J., Marocco, A. & Munkvold, G. P. Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genomics 16, 1089 (2015).
Liu, F., Wu, J., Zhan, R. & Ou, X. Transcription profiling analysis of mango – Fusarium mangiferae interaction. Front. Microbiol. 7, 1443 (2016).
Zuluaga, A. P. et al. Transcriptome responses to Ralstonia solanacearum infection in the roots of the wild potato Solanum commersonii. BMC Genomics 16, 246 (2015).
Kong, L. et al. Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genomics 16, 801 (2015).
Sangwan, R. S., Tripathi, S., Singh, J., Narnoliya, L. K. & De Sangwan, N. S. novo sequencing and assembly of Centella asiatica leaf transcriptome for map** of structural, functional and regulatory genes with special reference to secondary metabolism. Gene 525, 58–76 (2013).
Cho, W. K., Lian, S., Kim, S., Seo, B. Y. & Jung, J. K. Time-course RNA-Seq analysis reveals transcriptional changes in rice plants triggered by rice stripe virus infection. PLoS ONE 10, e0136736. https://doi.org/10.1371/journal.pone.0136736 (2015).
Muhae-Ud-Din, G., Chen, D., Liu, T., Chen, W. & Gao, L. Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 10, 9029. https://doi.org/10.1038/s41598-020-65748-w (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L. & Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22 (2011).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Anders, S., Pyl, P. T. & Huber, W. Genome analysis HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Anders, S. & Huber, W. Differential expression of RNA-Seq data at the gene level – the DESeq package. Eur. Mol. Biol. Lab., 10, f1000research (2012).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, 480–484 (2008).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, 14 (2010).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29, e45–e45 (2001).
Sudisha, J., Sharathchandra, R. G., Amruthesh, K. N., Kumar, A. & Shetty, H. S. Pathogenesis related proteins in plant defense response. In Plant defence: biological control 379–403, (2012).
Sels, J., Mathys, J., De Coninck, B. M. A., Cammue, B. P. A. & De Bolle, M. F. C. Plant pathogenesis-related (PR) proteins: A focus on PR peptides. Plant Physiol. Biochem. 46, 941–950 (2008).
Ebrahim, S., Usha, K. & Singh, B. Pathogenesis related (PR) proteins in plant defense mechanism. Sci. Against Microb. Pathog. 2, 1043–1054 (2011).
Anguelova-Merhar, V. S., VanDer Westhuizen, A. J. & Pretorius, Z. A. β-1, 3-glucanase and chitinase activities and the resistance response of wheat to leaf rust. J. Phytopathol. 149, 381–384 (2001).
Han, Y. et al. Differential expression profiling of the early response to Ustilaginoidea virens between false smut resistant and susceptible rice varieties. BMC Genomics 16, 955 (2015).
Manghwar, H. et al. Expression analysis of defense related genes in wheat and maize against Bipolaris sorokiniana. Physiol. Mol. Plant Pathol. 103, 36–46 (2018).
Pandey, S. P. & Somssich, I. E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 150, 1648–1655 (2009).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Phukan, U. J., Jeena, G. S. & Shukla, R. K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 7, 760 (2016).
Bai, Y., Sunarti, S., Kissoudis, C., Visser, R. G. F. & van der Linden, C. G. The role of tomato WRKY genes in plant responses to combined abiotic and biotic stresses. Front. Plant Sci. 9, 801 (2018).
Tsuda, K. & Somssich, I. E. Transcriptional networks in plant immunity. New Phytol. 206, 932–947 (2015).
Grunewald, W. et al. A role for AtWRKY23 in feeding site establishment of plant-parasitic nematodes. Plant Physiol. 148, 358–368 (2008).
Murray, S. L., Ingle, R. A., Petersen, L. N. & Denby, K. J. Basal resistance against Pseudomonas syringae in Arabidopsis involves WRKY53 and a protein with homology to a nematode resistance protein. Mol. Plant Microbe Interact. 20, 1431–1438 (2007).
Mukhtar, M. S., Deslandes, L., Auriac, M. C., Marco, Y. & Somssich, I. E. The Arabidopsis transcription factor WRKY27 influences wilt disease symptom development caused by Ralstonia solanacearum. Plant J. 56, 935–947 (2008).
Wang, X. et al. Ectopic expression of the wild grape WRKY transcription factor vqWRKY52 in Arabidopsis thaliana enhances resistance to the biotrophic pathogen powdery mildew but not to the necrotrophic pathogen Botrytis cinerea. Front. Plant Sci. 8, 97 (2017).
Gish, L. A. & Clark, S. E. The RLK/Pelle family of kinases. Plant J. 66, 117–127. https://doi.org/10.1111/j.1365-313X.2011.04518.x (2011).
Ghareeb, H., Becker, A., Iven, T., Feussner, I. & Schirawski, J. Sporisorium reilianum infection changes inflorescence and branching architectures of maize. Plant Physiol. 156, 2037–2052 (2011).
Shiu, S. & Bleecker, A. B. Plant receptor-like kinase gene family: diversity, function, and signaling. Sci. STKE. 113, 22 (2001).
Hosseini, S., Elfstrand, M., Heyman, F., Jensen, D. F. & Karlsson, M. Deciphering common and specific transcriptional immune responses in pea towards the oomycete pathogens Aphanomyces euteiches and Phytophthora pisi. BMC Genomics 16, 627 (2015).
Andreasson, E. et al. The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J. 24, 2579–2589 (2005).
Berger, S. et al. Visualization of dynamics of plant-pathogen interaction by novel combination of chlorophyll fluorescence imaging and statistical analysis: differential effects of virulent and avirulent strains of P. syringae and of oxylipins on A. thaliana. J. Exp. Bot. 58, 797–806 (2007).
Ehness, R., Ecker, M., Godt, D. E. & Roitsch, T. Glucose and stress independently regulate source and sink metabolism and defense mechanisms via signal transduction pathways involving protein phosphorylation. Plant Cell 9, 1825–1841 (1997).
Berger, S., Sinha, A. K. & Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 58, 4019–4026 (2007).
Siemens, J. et al. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant-Microbe Interact. 19, 480–494 (2006).
Carroll, C. S. & Moore, M. M. Ironing out siderophore biosynthesis: a review of non-ribosomal peptide synthetase (NRPS)-independent siderophore synthetases. Crit. Rev. Biochem. Mol. Biol. 53, 356–381 (2018).
Kadi, N. & Challis, G. L. Siderophore biosynthesis. a substrate specificity assay for nonribosomal peptide synthetase-independent siderophore synthetases involving trap** of acyl-adenylate intermediates with hydroxylamine. Meth. Enzymol. 458, 431–456 (2009).
Schröder, I., Johnson, E. & De Vries, S. Microbial ferric iron reductases. FEMS Microbiol. Rev. 27, 427–447 (2003).
Howard, D. H. Acquisition, transport, and storage of iron by pathogenic fungi. Clin. Microbiol. Rev. 12, 394–404 (1999).
Kronstad, J. W. & Caza, M. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell. Infect. Microbiol. 4, 80 (2013).
Nelson, D. R., Schuler, M. A., Paquette, S. M., Werck-Reichhart, D. & Bak, S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 135, 756–772 (2004).
Tanabe, S. et al. A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell 17, 776–790 (2005).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31761143011), the National Key Research and Development Programme of China (2018YFD0200406) and the Chinese Ministry of Agriculture (CARS-03).
Author information
Authors and Affiliations
Contributions
L.G. designed and wrote the article; Z.R., J.L., G.M., H.Z. and Z.D. performed the experiment; J.Z., S.Z., T.L. and W.C. provided the materials, and all authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, Z., Liu, J., Din, G.M.U. et al. Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci Rep 10, 21567 (2020). https://doi.org/10.1038/s41598-020-78628-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78628-0
- Springer Nature Limited