
Abstract
Previous transcriptome studies of the human endometrium have revealed hundreds of simultaneously up- and down-regulated genes that are involved in endometrial receptivity. However, the overlap between the studies is relatively small, and we are still searching for potential diagnostic biomarkers. Here we perform a meta-analysis of endometrial-receptivity associated genes on 164 endometrial samples (76 from ‘pre-receptive’ and 88 from mid-secretory, ‘receptive’ phase endometria) using a robust rank aggregation (RRA) method, followed by enrichment analysis, and regulatory microRNA prediction. We identify a meta-signature of endometrial receptivity involving 57 mRNA genes as putative receptivity markers, where 39 of these we confirm experimentally using RNA-sequencing method in two separate datasets. The meta-signature genes highlight the importance of immune responses, the complement cascade pathway and the involvement of exosomes in mid-secretory endometrial functions. Bioinformatic prediction identifies 348 microRNAs that could regulate 30 endometrial-receptivity associated genes, and we confirm experimentally the decreased expression of 19 microRNAs with 11 corresponding up-regulated meta-signature genes in our validation experiments. The 57 identified meta-signature genes and involved pathways, together with their regulatory microRNAs could serve as promising and sought-after biomarkers of endometrial receptivity, fertility and infertility.
Similar content being viewed by others
Introduction
The period of endometrial receptivity, also known as the window of implantation (WOI), is the limited time (one to two days) when luminal epithelium is favourable for embryo adhesion as the first step of implantation1. Successful embryo implantation depends on synchronization of a viable embryo and receptive endometrium. In fact, inadequate uterine receptivity has been estimated to contribute to one third of implantation failures, whereas the embryo itself is responsible for two thirds of them2, 3. In assisted reproductive technologies where good-quality embryos are transferred as a standard of care, implantation failure remains an unsolved obstacle4,5,6. In patients with recurrent implantation failure (RIF) temporal displacement of the WOI has been described in one out of four patients7, thus suggesting the possibility of these women suffering RIF of endometrial origin. Further, impaired decidualization of endometrial stromal cells that predisposes to late implantation may negate endometrial ‘embryo quality control’ and cause early pregnancy failure8,9,10. Hence, better understanding of endometrial receptivity and the importance of the mechanisms involved in mid-secretory endometrial functions is warranted.
From the first histological dating methods11, 12 to the new ‘omics’ technologies, extensive efforts have been made to understand and characterise receptive endometrium. Traditional endometrial dating criteria, like tissue histology, are obsolete, since their accuracy, reproducibility and functional relevance have been questioned in various randomised studies13, 14. This has encouraged further investigation and application of new technologies to diagnose endometrial receptivity objectively, since reliable diagnostic markers are still lacking and the molecular mechanisms remain largely unclear15,16,17.
With the ‘omics’ revolution, the quest for the transcriptomic signature of human endometrial receptivity has revealed hundreds of simultaneously up- and down-regulated genes implicated in the phenomenon (reviewed in ref. 18). While any given study yields a number of genes, the overlap between different studies is relatively small. The perceived limitations of this technology have been well defined and lie in differences in experimental design, timing and conditions of endometrial sampling, selection criteria regarding patients, transcriptome array/sequencing platforms and genome annotation versions used, pipelines for data processing and a lack of consistent standards for data presentation19,20,21,22,23.
To overcome the aforementioned limitations in endometrial transcriptome analyses, we applied a recently published robust rank aggregation (RRA) method24, followed by enrichment analysis, to identify a meta-signature or consensus signature of highly putative biomarkers of endometrial receptivity. Additionally, we set up to analyse possible microRNAs that could influence the endometrial receptivity-associated genes/mRNAs. Further, we aimed to experimentally validate the meta-signature mRNA genes and their regulatory miRNAs in two independent sample sets.
Results
Identification of relevant studies
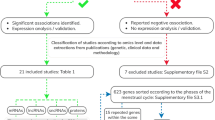
The search process and results of the systematic literature review are presented in detail in Supplementary Figure 1. Eventually, out of 57 eligible publications, 14 remained suitable for qualitative analysis. Five eligible studies25,26,27,28,29 were not included in the final analysis, since the data on lists of differentially expressed genes were not available publicly nor in response to requests to the authors. A detailed description of the studies included in the final analysis is presented in Table 1. Our pooled dataset obtained from the nine remaining studies covered 76 ‘pre-receptive’-phase (28 biopsy samples from the proliferative phase and 48 from the early secretory phase) and 88 mid-secretory, ‘receptive’ phase endometrial samples.
Meta-signature of endometrial receptivity-associated genes
Using robust rank aggregation analysis, we identified a statistically significant meta-signature of 52 up-regulated and five down-regulated genes in mid-secretory vs. ‘pre-receptive’ endometrium (see Table 2). The up-regulated transcripts with the highest scores in receptive-phase endometrium were PAEP, SPP1, GPX3, MAOA and GADD45A. The five down-regulated transcripts identified as receptivity-associated genes were SFRP4, EDN3, OLFM1, CRABP2 and MMP7.
Enrichment analyses
We used up-to-date enrichment analysis software (g:Profiler) for analysis of biological processes and pathways connected to the meta-signature of mid-secretory endometrium. A significant proportion of the genes were involved in biological processes such as responses to external stimuli, responses to wounding, inflammatory responses, negative regulation of coagulation, humoral immune responses, and immunoglobulin-mediated immune responses, among others. Fig. 1 and Supplementary Table 1 show the connections of the 57 endometrial receptivity genes with their respective Gene Ontology biological processes. The only significantly enriched pathway related to the meta-signature genes was a KEGG pathway of complement and coagulation cascades, where the identified genes were connected to the complement cascade part (p = 0.00112) (see Fig. 2). A significant number of the genes were also connected with the extracellular region and exosomes. In order to confirm the involvement of exosomes, we searched for the presence of the meta-signature genes in human exosomes based on the exosome database, ExoCarta (exocarta.org). Fisher’s Exact Test was performed to analyse if meta-signature genes were over represented in the exosome database. All the human protein coding genes were downloading from ENSEMBL v75 database (version February 2014) and mRNAs or proteins from Exocarta database (exocarta.org). Altogether, meta-signature genes had 2.13 times higher probability to be in the exosomes than the rest of the protein-coding genes in the human genome (Fisher’s exact test, two-sided p = 0.0059). The 28 identified proteins from the meta-signature gene list that have been shown to be in exosomes are presented in Fig. 3 that illustrates the involvement of extracellular vesicles (exosomes and microvesicles) in embryo implantation process.

Gene ontology (GO) processes and the pathways most strongly enriched among endometrial receptivity-associated genes. Genes are presented on the right side on the circle and the correlating GO processes, cellular compartments and pathways are on the left side.

Schematic overview of the 57 meta-signature genes, their literature-based localisation and involvement in the mid-secretory phase endometrium. Different membrane-associated proteins (ABCC3, ANXA2, ANXA4, AQP3, CD55, DKK1, DPP4, EDN3, EDNRB, EFNA1, ENPEP, SFRP4, SLC1A1, SPP1, TSPAN8), epithelial cell tight junction protein (CLDN4), secreted enzymes and binding proteins (APOD, CP, GPX3, IGFBP1, TCN1), secreted immune response proteins (DEFB1, GLNY, IL15, PAEP), extracellular matrix-associated proteins (COMP, HABP2, LAMB3, MMP7), different enzymes (ACADSB, AOX1, ARG2, IDO1, MAOA, NNMT), signalling proteins (C10orf10, GBP2, G0S2, MAP3K5, NDRG1), metallothioneins (MT1G, MT1H), DNA binding and repair proteins (ARID5B, DDX52, GADD45A), transcription factors (BCL6, CEBPD, ID4), and other intracellular proteins (CRABP2, DYNLT3, OLFM1, PRUNE2, S100P) are indicated. Additionally, the enriched KEGG pathway of complement cascade with the identified genes C1R, SERPING1, CD55, C4BPA and CFD is highlighted. (Figure created by Elsevier Illustration Service).

Extracellular vesicles (EVs) – exosomes and microvesicles, in embryo-endometrium cross-talk. In the exosomes the meta-signature genes are highlighted (based on ExoCarta database). Exosomes (30–100 nm) are generated from inward budding of the endosomal membrane, resulting in formation of a multivesicular body. Microvesicles (100 nm–1 μm) are produced by direct budding of the plasma membrane. Membrane-associated (bubbles) and transmembrane proteins (cylinders), and nucleic acids (DNA, RNA, curved symbols) are selectively incorporated into the EVs. EVs may dock on the plasma membrane of a target cell (A), fuse directly with the plasma membrane (B), or be endocytosed (C). Endocytosed vesicles may subsequently fuse with the delimiting membrane of an endocytic compartment (D). Both (B and D) pathways result in the delivery of proteins and nucleic acids into the membrane or cytosol of the target cell. (Figure adapted with permission from62, 84, created by Elsevier Illustration Service).
Validation of meta-signature genes in two independent sample sets
Meta-analysis identified 57 genes differentially expressed between the ‘pre-receptive’ and mid-secretory endometrium, where 52 genes were up- and five were down-regulated at WOI. Our RNA-sequencing (RNA-seq) analysis on 20 independent endometrial biopsy samples from fertile women confirmed the differential expression of 52 meta-signature genes (all of them with fold change of ≥3) – 48 of these genes were likewise up-regulated and four were down-regulated (CRABP2, EDN3, OLFM1, SFRP4) in the mid-secretory endometria (Fig. 4). MMP7 and CFD were not differentially expressed in our RNA-seq analysis of LH+8 vs. LH+2 phase endometria. Three genes, COMP, MT1H, S100P, did not pass the initial filtering of RNA-seq data (counts per million, CPM > 2 in at least 15 samples), which might be due to their low expression levels. The filtering was applied to rule out transcripts with very low or inconsistent expression levels across individuals.

Validation of the meta-signature genes in two independent sample sets. RNA-seq analysis of endometrial tissue samples confirmed differential expression of 52 (91.2%) meta-signature genes in the mid-secretory phase endometrium vs. early secretory phase endometrium. Cell type-specific RNA-seq analysis of endometrial epithelial and stromal cells confirmed differential expression of 43 (75.4%) meta-signature genes in those cell populations in the mid-secretory endometrium vs. early secretory endometrium. In total, 39 (68.4%) meta-signature genes (typed in white colour) were identified in validation experiments on two different sample sets, where 35 genes were up-regulated and 4 genes (CRABP2, EDN3, OLFM1, SFRP4) down-regulated in the mid-secretory phase endometrium.
Next, we investigated the expression of the 57 meta-signature genes in FACS (fluorescence-activated cell sorting)-sorted endometrial epithelial and stromal cells from two time points in the menstrual cycle, early secretory vs. mid-secretory phase, from 16 fertile women. Thirty-nine of those genes were significantly up-regulated and four were down-regulated (CRABP2, EDN3, OLFM1, SFRP4) in the receptive phase in those cell populations (all of them with fold change of ≥2) (Fig. 4; Supplementary Figure 2). Although most of the genes were up-regulated in both cell types, it is notable that the expression of ANXA2, COMP, CP, DDX52, DPP4, DYNLT3, EDNRB, EFNA1, G0S2, HABP2, LAMB3, MAOA, NDRG1, PRUNE2, SPP1, and TSPAN8 was epithelium-specific (Fig. 5), while none of the genes was down-regulated in the epithelial cells only. The stroma-specific up-regulated genes were APOD, CFD, C1R and DKK1, and down-regulated gene was OLFM1 (Fig. 5). It is noteworthy that although most of the genes were up-regulated in both cell types, the expression of these genes was still higher in the epithelial cells.

Validation of the meta-signature genes on cell type-specific RNA-seq data. Significantly up-regulated (orange) and down-regulated (blue) genes in FACS-sorted stromal and epithelial cells. The x-scale represents log2(FC) between LH+8 vs. LH+2 comparisons in stromal and epithelial cells. When comparing the gene expression values between epithelial vs. stromal cells in the mid-secretory phase endometrium (LH+8), most genes were more up-regulated in the epithelial cells (higher expression highlighted as darker orange). All reported results are significant at FDR < 0.05.
Further validation of these confirmed meta-signature genes was carried out with real-time PCR. Up-regulation of DDX52, DYNLT3, C1R and APOD expression levels in the receptive phase endometrial samples was confirmed (Supplementary Figure 3). Furthermore, the cell-specific up-regulated DDX52 and DYNLT3 expression was confirmed in FACS-sorted epithelial cells, and the stromal cell-specific C1R and APOD up-regulation was confirmed in the FACS-sorted stromal cells (Supplementary Figure 3).
In conclusion, the validation of the 57 meta-analysis consensus genes of the receptive phase endometrium among the two independent sets of endometrial tissue samples and cell-populations analysed confirmed the differential expression of 39 genes, with 35 up- and 4 down-regulated expression during WOI (Fig. 4).
In silico analysis of potential microRNAs regulating meta-signature genes
To evaluate the potential regulation of the 57 meta-signature genes, we predicted their putative regulatory-microRNAs using three different in silico target prediction algorithms. DIANA microT-CDS predicted 1,355 microRNAs with 12,627 potential binding sites, TargetScan 7.0 predicted 2,521 microRNAs with 32,560 potential binding sites, and miRanda predicted 2,568 microRNAs with 42,413 potential binding sites. The overlap between all three algorithms resulted in 818 microRNAs and 1,403 potential unique binding sites for 43 meta-signature genes (Supplementary Table 2).
To add an additional filter to the bioinformatic predictions, we overlaid those with experimentally determined Argonaute binding sites (microRNAs regulate gene expression by guiding Argonaute proteins to specific target mRNA sequences), mined from publicly available AGO-CLIP datasets. Out of 1,403 intersected potential binding sites, 395 showed overlap with experimentally determined Argonaute binding site in human cell lines, filtering down to the most probable microRNA and mRNA interactions. These 395 sites included interactions between 30 genes from our original meta-signature gene list and 348 microRNAs (Supplementary Table 3).
Validation analysis of predicted microRNAs and target mRNAs in independent sample set
In silico analysis of potential microRNAs regulating the meta-signature genes predicted interactions between 30 meta-signature genes and 348 microRNAs. Using the list of the predicted interactions, we investigated if these potentially interacting microRNAs are significantly regulated in our endometrial microRNA-sequencing data on endometrial biopsies from the mid-secretory phase vs. early secretory phase of healthy fertile women. We identified 19 microRNAs that were significantly down-regulated in the mid-secretory endometria with corresponding 11 meta-signature genes to be significantly up-regulated in our sample set (Fig. 6). Based on the TargetScan context++ scores, the probability of the interaction between microRNA and its target gene seems to be higher in pairs miR-449c-5p and DKK1, miR-450b-5p, miR-424-5p, miR-130b-3p and IL15, miR-500a-5p and GADD45A, and miR-181a-2-3p and ACADSB. When focussing only on the genes that were confirmed in both independent validation analyses (RNA-seq of endometrial biopsies and cell type-specific RNA-seq), five target genes (ANXA4, ARID5B, DKK1, EFNA1 and SPP1) and 10 microRNAs remained important (Fig. 6).

In silico predicted interactions between significantly up-regulated mRNAs (red) and down-regulated microRNAs (green) in LH+8 vs. LH+2 endometrium. The colour intensity indicates the strength of up- or down-regulation (FDR < 0.05). The colour of the arrows between the microRNA and mRNA represents TargetScan context++ score (see Supplementary Table 2 for scores), where darker arrow shows more probable interaction. The number of arrows between microRNA and mRNA indicates different microRNA binding sites within the same transcript. Meta-signature genes that were confirmed in both independent validation analyses together with their corresponding miRNAs are highlighted with black circle/diamond borders.
Discussion
In this report, we present a systematic review and meta-analysis approach together with comprehensive experimental validation in order to identify promising biomarkers and molecular pathways involved in mid-secretory endometrial functions. Analysing the lists of differentially expressed genes from previously published expression profiling studies, we established a meta-signature of receptive endometrium with 57 genes as putative receptivity biomarkers. Interestingly, the commercial transcriptome-based endometrial receptivity diagnostic tool ERA (Endometrial Receptivity Array)7, 30 shares 47 genes in common with the identified meta-signature. Validation of the meta-signature genes in two different sample sets of healthy fertile women in mid-secretory vs. early secretory endometria using the up-to-date transcriptome analysis by RNA-seq confirmed 39 meta-signature genes.
The human endometrial transcriptome has been extensively studied in the past decade in a search of identifying diagnostic markers of receptive endometrium and to provide more understanding into the complex regulation of endometrial functions. Despite of the mass ‘omics’ data generated, only three in silico data-mining studies17, 31, 32 using previously published gene expression data have been published to date. Bhagwat et al. created a Human Gene Expression Endometrial Receptivity database (HGEx-ERdb) of 19,285 genes expressed in human endometrium, among which they identified 179 receptivity-associated genes32. Zhang et al. analysed raw data from three previous microarray studies33,34,35 and proposed 148 potential biomarkers of receptive endometrium17, while Tapia et al. integrated gene lists from seven previous microarray studies and presented a list of 61 endometrial receptivity biomarkers31. These three in silico analysis studies share only nine genes in common, highlighting the differences not only in in silico analysis approaches applied but also the great variation in study designs, analysis methods and data processing in published transcriptome studies. Clearly the mass of data generated within endometrial transcriptomics studies is under-explored, challenging investigators in future to analyse huge sets of data simultaneously in order to raise power, credibility and reliability of the findings.
The preferred method for gene expression meta-analysis requires analysis of raw expression datasets. However, such a thorough analysis is often not possible as a result of unavailability of raw data, which is partially the case in our meta-analysis. Variation in the number of gene transcripts known at a given moment together with the technological platform employed makes proper integration of raw datasets complicated. In addition, the limited sample size and noisiness of microarray data have resulted in inconsistency of biological conclusions36. In order to overcome these limitations, we directly analysed lists of differentially expressed genes from nine published studies involving a total of 164 endometrial biopsy samples from healthy women. Using a method that has been specifically designed for comparison of gene lists and identification of commonly overlap** genes in various studies, including recently published transcriptome studies in different ethnic groups30, 37, 38, we hope to provide an up-to-date meta-signature of endometrial receptivity biomarkers. Nevertheless, we have to bear in mind that with our approach, analysing the significantly differentially expressed gene lists, we could have missed the potential biomarker genes that were below statistical significance in individual studies but could become relevant in a meta-analysis.
The 57 genes identified in our meta-analysis could serve as the top-priority biomarkers of receptive phase endometrium in humans. Of special interest is SPP1, which was detected in all transcriptome studies that were included in our meta-analysis, together with ANXA4, CLDN4, DPP4, GPX3, MAOA, and PAEP, as they have also been identified as putative biomarkers of endometrial receptivity in the previous data-mining and review studies17, 19, 29, 31, 32.
Secreted phosphoprotein 1, SPP1, also known as osteopontin, is a secreted extracellular matrix (ECM) protein that binds to different cell-surface integrins to stimulate cell–cell and cell–ECM adhesion and communication (see Fig. 2), which play a part in the implantation process in various species39,40,41. It is generally accepted that SPP1 interacts with apically expressed integrins on the luminal endometrial epithelium and embryo trophectoderm to attach the conceptus to the endometrium39. Indeed, our cell type-specific RNA-seq validation analysis of endometrial epithelial and stromal cells demonstrates that SPP1 is up-regulated only in the epithelial cells (though in our setting we had a mixture of both luminal and glandular epithelial cells) and not in the stromal cells in the receptive phase endometrium (Fig. 5). Dysregulation of osteopontin in mid-secretory endometria of women with various reproductive disorders has been detected in several studies42,43,44,45,33, the differentially expressed probe lists were acquired by reanalysing the data stored in the Gene Expression Omnibus under accession number GSE6364. We used GEO2R web tool (http://www.ncbi.nlm.nih.gov/geo/info/geo2r.html) with default options for differential analysis and gene list acquisition (false discovery rate, FDR < 0.05; fold change, FC > 2.0). Probes not annotated by ENTREZ IDs were removed from subsequent analyses.
Subsequently, lists of up- and down-regulated genes were ranked by their fold changes. In cases of multiple probes detecting the same gene, only the probe with the largest absolute fold change was used for list construction.
Meta-analysis
The robust rank aggregation algorithm (RRA package v.1.1) was used for meta-analysis of the ranked gene lists24. To assess full gene list sizes, the number of detectable gene ENTREZ IDs was used for each array platform and all ENTREZ IDs from BioMart v.75 were used for the RNA-seq study38. For correcting for multiple testing, we used a strict Bonferroni threshold by multiplying all P-values by the maximal number of elements in all input lists. In our case this involved the data published by Hu et al.38, where the total number of ENTREZ IDs available in ENSEMBL v.75 (39,030) was used as the total number of tests. All lists used in the analysis reflect expression in the mid-secretory group compared with another group (proliferative or early-secretory phase).
Enrichment analysis
Enrichment analyses for Gene Ontology (GO) terms and biological pathways (KEGG and Reactome) were carried out by using the g:Profiler web tool (biit.cs.ut.ee/gprofiler/)76, 77. This software was chosen over other enrichment analysis tools as it is up-to-date (updated in May 2016 to Ensembl 84 and Ensembl Genomes 31) and it provides a compact graphical output. The obtained results were corrected for multiple testing by using the g:Profiler tailor-made algorithm g:SCS, which has been shown to provide a better threshold between significant and non-significant results than (commonly used) Bonferroni correction or the Benjamini-Hochberg false discovery rate76.
In silico analysis of potential microRNAs regulating meta-signature genes
MicroRNA target prediction was performed using three different algorithms – DIANA microT-CDS, TargetScan 7.0 and miRanda v3.3a. In DIANA microT-CDS78 and TargetScan 7.079 precomputed prediction results were downloaded from their respective websites (diana.imis.athena-innovation.gr/DianaTools/index.php?r = microT_CDS/index; www.targetscan.org/cgi-bin/targetscan/data_download.cgi?db = vert_70). DIANA microT-CDS utilises ENSEMBL v69 transcriptome and miRBase v18 for the prediction, whereas TargetScan 7.0 uses ENSEMBL v75 and miRBase v2180. miRanda v3.3a binary81 was downloaded from http://www.microrna.org/microrna/getDownloads.do and used for the target prediction with ENSEMBL v75 3′UTRs and miRBase v21 mature sequences as an input. The algorithm was used with default settings: Gap Open Penalty: −9.0, Gap Extend Penalty: −4.0, Score Threshold: 140.0, Energy Threshold: 1.0 kcal/mol and Scaling Parameter: 4.0. miRBase internal IDs were used to standardise microRNA names between different miRBase versions.
For additional support to in silico target predictions, we used database harbouring experimental data about mammalian microRNA binding sites. Therefore, Argonaute (AGO1, AGO2, AGO3 and AGO4) HITS-CLIP and PAR-CLIP datasets for human cell lines were downloaded from StarBase v.2.082 website in the BED format and overlaid with predicted microRNA target sites (http://starbase.sysu.edu.cn/download.php).
Validation of meta-signature genes and predicted microRNAs in the independent sample sets
We validated the meta-signature genes in our two independent sample sets from NOTED project (EU-FP7 Eurostars Programme, EU41564) and SARM project (EU-FP7, IAPP, EU324509). The studies were approved by the local Research Ethics Committees of the University of Tartu and Instituto Valenciano de Infertilidad. An informed consent was signed by all women who agreed to participate in the study, and all methods were carried out in accordance with relevant guidelines and regulations.
The detailed description of the study participants and RNA-seq analysis within NOTED project is described in the Supplementary Material. Briefly, 20 healthy fertile women provided endometrial biopsy samples in the early secretory phase (2 days after the luteinizing hormone (LH) peak, LH+2) and in the mid-secretory phase of the menstrual cycle (LH+8) within the same natural cycle. Total mRNA transcriptome and microRNA profile analysis from the same biopsy samples were performed with the RNA-seq method. Differential expression was tested using the edgeR statistical package. Up-regulation was defined as statistically significantly (FDR corrected p-value < 0.05) higher expression (expressed as ‘counts per million reads’, CPM) in the mid-secretory phase samples, whereas down-regulation was defined as statistically significantly lower expression in the mid-secretory samples. The primary RNA-seq data are available in the public database Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) under accession number GSE98386.
The other independent set of validation was carried out in additional 16 healthy fertile women within SARM project, where cell type-specific RNA-seq on endometrial samples was performed, with separated epithelial and stromal cells. Briefly, endometrial biopsies were collected from 16 healthy fertile women on two different time points within the natural cycle, LH+2 and LH+8. Single cells from endometrial biopsy samples were separated, epithelial cells were labelled with fluorochrome-conjugated mouse anti-human CD9 monoclonal antibody and stromal cells were simultaneously labelled with fluorochrome-conjugated mouse anti-human CD13 monoclonal antibody, followed by flow cytometric analysis and FACS cell sorting. Bulk-RNA full transcriptome analysis of FACS sorted endometrial epithelial and stromal cells was performed with the RNA-seq method, following the single-cell tagged reverse transcription (STRT) protocol with modifications83. Differential expression was tested using the edgeR statistical package. Up-regulation was defined as statistically significantly (FDR corrected p-value < 0.05) higher expression (expressed as ‘normalised read counts’) in the mid-secretory phase samples, whereas down-regulation was defined as statistically significantly lower expression in the mid-secretory samples. The primary RNA-seq data are available in the public database Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) under accession number GSE97929. Detailed protocol of the sample collection, processing, and analysis is described in the Supplementary Material.
Further validation of the endometrial receptivity signature genes DDX52, DYNLT3, C1R and APOD on NOTED and SARM project samples using real-time PCR is described in the Supplementary Material.
References
Wilcox, A. J., Baird, D. D. & Weinberg, C. R. Time of implantation of the conceptus and loss of pregnancy. N Engl J Med 340, 1796–1799 (1999).
Macklon, N. S., Stouffer, R. L., Giudice, L. C. & Fauser, B. C. The science behind 25 years of ovarian stimulation for in vitro fertilization. Endocr Rev 27, 170–207 (2006).
Cha, J., Vilella, F., Dey, S. & Simón, C. In Ten Critical Topics in Reproductive Medicine 44–48 (Science/AAAS, Washington DC, 2013).
Edwards, R. G. Clinical approaches to increasing uterine receptivity during human implantation. Hum Reprod 10(Suppl 2), 60–66 (1995).
Margalioth, E. J., Ben-Chetrit, A., Gal, M. & Eldar-Geva, T. Investigation and treatment of repeated implantation failure following IVF-ET. Hum. Reprod. 21, 3036–43 (2006).
Simon, A. & Laufer, N. Repeated implantation failure: clinical approach. Fertil. Steril. 97, 1039–43 (2012).
Ruiz-Alonso, M. et al. The endometrial receptivity array as diagnosis and personalized embryo transfer as treatment for patients with receptive implantation failure. Fertil Steril in press (2013).
Teklenburg, G., Salker, M., Heijnen, C., Macklon, N. S. & Brosens, J. J. The molecular basis of recurrent pregnancy loss: impaired natural embryo selection. Mol Hum Reprod 16, 886–895 (2010).
Teklenburg, G. et al. Natural selection of human embryos: decidualizing endometrial stromal cells serve as sensors of embryo quality upon implantation. PLoS One 5, e10258 (2010).
Salker, M. S. et al. Disordered IL-33/ST2 activation in decidualizing stromal cells prolongs uterine receptivity in women with recurrent pregnancy loss. PLoS One 7, e52252 (2012).
Noyes, R. W., Hertig, A. T. & Rock, J. Dating the endometrial biopsy. Fertil Steril 1, 3–25 (1950).
Noyes, R. W., Hertig, A. T. & Rock, J. Dating the endometrial biopsy. Am J Obs. Gynecol 122, 262–263 (1975).
Coutifaris, C. et al. Histological dating of timed endometrial biopsy tissue is not related to fertility status. Fertil. Steril. 82, 1264–1272 (2004).
Murray, M. J. et al. A critical analysis of the accuracy, reproducibility, and clinical utility of histologic endometrial dating in fertile women. Fertil. Steril. 81, 1333–1343 (2004).
Brinsden, P. R., Alam, V., de Moustier, B. & Engrand, P. Recombinant human leukemia inhibitory factor does not improve implantation and pregnancy outcomes after assisted reproductive techniques in women with recurrent unexplained implantation failure. Fertil Steril 91, 1445–1447 (2009).
Lessey, B. A. Assessment of endometrial receptivity. Fertil. Steril. 96, 522–529 (2011).
Zhang, D., Sun, C., Ma, C., Dai, H. & Zhang, W. Data mining of spatial-temporal expression of genes in the human endometrium during the window of implantation. Reprod. Sci. 19, 1085–1098 (2012).
Altmäe, S. et al. Guidelines for the design, analysis and interpretation of ‘omics’ data: focus on human endometrium. Hum. Reprod. Update 20, 12–28 (2014).
Horcajadas, J. A., Pellicer, A. & Simon, C. Wide genomic analysis of human endometrial receptivity: new times, new opportunities. Hum Reprod Updat. 13, 77–86 (2007).
Altmäe, S. et al. Endometrial gene expression analysis at the time of embryo implantation in women with unexplained infertility. Mol Hum Reprod 16, 178–187 (2010).
Ruiz-Alonso, M., Blesa, D. & Simon, C. The genomics of the human endometrium. Biochim. Biophys. Acta 1822, 1931–1942 (2012).
Ulbrich, S. E., Groebner, A. E. & Bauersachs, S. Transcriptional profiling to address molecular determinants of endometrial receptivity–lessons from studies in livestock species. Methods 59, 108–115 (2013).
Aghajanova, L., Hamilton, A. E. & Giudice, L. C. Uterine receptivity to human embryonic implantation: histology, biomarkers, and transcriptomics. Semin Cell Dev Biol 19, 204–211 (2008).
Kolde, R., Laur, S., Adler, P. & Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28, 573–80 (2012).
Otsuka, A. Y., Andrade, P. M., Villanova, F. E., Borra, R. C. & Silva, I. D. C. G. Human endometrium mRNA profile assessed by oligonucleotide three-dimensional microarray. Gynecol. Endocrinol. 23, 527–34 (2007).
Haouzi, D. et al. Identification of new biomarkers of human endometrial receptivity in the natural cycle. Hum Reprod 24, 198–205 (2009).
Ponnampalam, A. P., Weston, G. C., Trajstman, A. C., Susil, B. & Rogers, P. A. W. Molecular classification of human endometrial cycle stages by transcriptional profiling. Mol. Hum. Reprod. 10, 879–93 (2004).
Tong, X.-M., Lin, X.-N., Song, T., Liu, L. & Zhang, S. Calcium-binding protein S100P is highly expressed during the implantation window in human endometrium. Fertil. Steril. 94, 1510–8 (2010).
Tseng, L. H. et al. Genome-based expression profiling as a single standardized microarray platform for the diagnosis of endometrial disorder: an array of 126-gene model. Fertil Steril 94, 114–119 (2010).
Diaz-Gimeno, P. et al. A genomic diagnostic tool for human endometrial receptivity based on the transcriptomic signature. Fertil Steril 95(50–60), 60–15 (2011).
Tapia, A., Vilos, C., Marin, J. C., Croxatto, H. B. & Devoto, L. Bioinformatic detection of E47, E2F1 and SREBP1 transcription factors as potential regulators of genes associated to acquisition of endometrial receptivity. Reprod. Biol. Endocrinol. 9, 14 (2011).
Bhagwat, S. R. et al. Endometrial receptivity: a revisit to functional genomics studies on human endometrium and creation of HGEx-ERdb. PLoS One 8, e58419 (2013).
Talbi, S. et al. Molecular phenoty** of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology 147, 1097–1121 (2006).
Burney, R. O. et al. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology 148, 3814–3826 (2007).
Hever, A. et al. Human endometriosis is associated with plasma cells and overexpression of B lymphocyte stimulator. Proc. Natl. Acad. Sci. USA 104, 12451–6 (2007).
Võsa, U. et al. Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 132, 2884–2893 (2013).
Altmäe, S. et al. Research resource: interactome of human embryo implantation: identification of gene expression pathways, regulation, and integrated regulatory networks. Mol. Endocrinol. 26, 203–217 (2012).
Hu, S. et al. Transcriptomic changes during the pre-receptive to receptive transition in human endometrium detected by RNA-Seq. J. Clin. Endocrinol. Metab. 99, E2744–53 (2014).
Johnson, G. A., Burghardt, R. C. & Bazer, F. W. Osteopontin: a leading candidate adhesion molecule for implantation in pigs and sheep. J. Anim. Sci. Biotechnol. 5, 56 (2014).
Kang, Y.-J., Forbes, K., Carver, J. & Aplin, J. D. The role of the osteopontin-integrin αvβ3 interaction at implantation: functional analysis using three different in vitro models. Hum. Reprod. 29, 739–49 (2014).
Liu, N., Zhou, C., Chen, Y. & Zhao, J. The involvement of osteopontin and β3 integrin in implantation and endometrial receptivity in an early mouse pregnancy model. Eur. J. Obstet. Gynecol. Reprod. Biol. 170, 171–6 (2013).
DuQuesnay, R. et al. Infertile women with isolated polycystic ovaries are deficient in endometrial expression of osteopontin but not alphavbeta3 integrin during the implantation window. Fertil. Steril. 91, 489–99 (2009).
Casals, G. et al. Osteopontin and alphavbeta3 integrin as markers of endometrial receptivity: the effect of different hormone therapies. Reprod. Biomed. Online 21, 349–59 (2010).
Casals, G. et al. Expression pattern of osteopontin and αvβ3 integrin during the implantation window in infertile patients with early stages of endometriosis. Hum. Reprod. 27, 805–13 (2012).
D’Amico, F. et al. Expression and localisation of osteopontin and prominin-1 (CD133) in patients with endometriosis. Int. J. Mol. Med. 31, 1011–6 (2013).
**ao, Y. et al. Expression of integrin β3 and osteopontin in the eutopic endometrium of adenomyosis during the implantation window. Eur. J. Obstet. Gynecol. Reprod. Biol. 170, 419–22 (2013).
Giudice, L. C. Microarray expression profiling reveals candidate genes for human uterine receptivity. Am. J. Pharmacogenomics 4, 299–312 (2004).
Giudice, L. C. Application of functional genomics to primate endometrium: insights into biological processes. Reprod Biol Endocrinol 4(Suppl 1), S4 (2006).
Haller-Kikkatalo, K., Altmäe, S., Tagoma, A., Uibo, R. & Salumets, A. Autoimmune activation toward embryo implantation is rare in immune-privileged human endometrium. Semin. Reprod. Med. 32, 376–84 (2014).
Janeway, C. A. & Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002).
Franchi, A., Zaret, J., Zhang, X., Bocca, S. & Oehninger, S. Expression of immunomodulatory genes, their protein products and specific ligands/receptors during the window of implantation in the human endometrium. Mol. Hum. Reprod. 14, 413–421 (2008).
Francis, J. et al. Impaired expression of endometrial differentiation markers and complement regulatory proteins in patients with recurrent pregnancy loss associated with antiphospholipid syndrome. Mol. Hum. Reprod. 12, 435–442 (2006).
Kao, L. C. et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology 144, 2870–2881 (2003).
Isaacson, K. B., Coutifaris, C., Garcia, C. R. & Lyttle, C. R. Production and secretion of complement component 3 by endometriotic tissue. J. Clin. Endocrinol. Metab. 69, 1003–9 (1989).
Tapia, A. et al. Differences in the endometrial transcript profile during the receptive period between women who were refractory to implantation and those who achieved pregnancy. Hum Reprod 23, 340–351 (2008).
Lee, J. et al. Differentially expressed genes implicated in unexplained recurrent spontaneous abortion. Int. J. Biochem. Cell Biol. 39, 2265–2277 (2007).
Ng, Y. H. et al. Endometrial exosomes/microvesicles in the uterine microenvironment: a new paradigm for embryo-endometrial cross talk at implantation. PLoS One 8, e58502 (2013).
Vilella, F. et al. Hsa-miR-30d, secreted by the human endometrium, is taken up by the pre-implantation embryo and might modify its transcriptome. Development 142, 3210–3221 (2015).
Machtinger, R., Laurent, L. C. & Baccarelli, A. A. Extracellular vesicles: roles in gamete maturation, fertilization and embryo implantation. Hum Reprod Updat, doi:10.1093/humupd/dmv055 (2015).
Saadeldin, M., Oh, H. & Lee, B. Embryonic – maternal cross-talk via exosomes: potential implications. Stem Cells Cloning Adv. Appl. 8, 103–107 (2015).
Evans, J. et al. Fertile ground: human endometrial programming and lessons in health and disease. Nat. Rev. Endocrinol, doi:10.1038/nrendo.2016.116 (2016).
Tannetta, D., Dragovic, R., Alyahyaei, Z. & Southcombe, J. Extracellular vesicles and reproduction-promotion of successful pregnancy. Cell. Mol. Immunol. 1–16, doi:10.1038/cmi.2014.42 (2014).
Burns, G. et al. Extracellular vesicles in luminal fluid of the ovine uterus. PLoS One 9, 15–19 (2014).
Luense, L. J., Carletti, M. Z. & Christenson, L. K. Role of Dicer in female fertility. Trends Endocrinol. Metab. 20, 265–72 (2009).
Carthew, R. W. & Sontheimer, E. J. Origins and Mechanisms of miRNAs and siRNAs. Cell 136, 642–655 (2009).
Hammond, S. M. An overview of microRNAs. Adv. Drug Deliv. Rev. 87, 3–14 (2015).
Lewis, B. P. et al. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005).
Sha, A. G. et al. Genome-wide identification of micro-ribonucleic acids associated with human endometrial receptivity in natural and stimulated cycles by deep sequencing. Fertil. Steril. 96, 150–155 e5 (2011).
Altmäe, S. et al. MicroRNAs miR-30b, miR-30d, and miR-494 Regulate Human Endometrial Receptivity. Reprod. Sci. 20, 308–317 (2013).
Kuokkanen, S. et al. Genomic profiling of microRNAs and messenger RNAs reveals hormonal regulation in microRNA expression in human endometrium. Biol. Reprod. 82, 791–801 (2010).
Boren, T. et al. MicroRNAs and their target messenger RNAs associated with endometrial carcinogenesis. Gynecol. Oncol. 110, 206–215 (2008).
Su, L. et al. Expression patterns of microRNAs in porcine endometrium and their potential roles in embryo implantation and placentation. PLoS One 9, e87867 (2014).
Chung, T. K. H. et al. Dysregulated microRNAs and their predicted targets associated with endometrioid endometrial adenocarcinoma in Hong Kong women. Int. J. cancer 124, 1358–65 (2009).
Wu, W., Lin, Z., Zhuang, Z. & Liang, X. Expression profile of mammalian microRNAs in endometrioid adenocarcinoma. Eur. J. Cancer Prev. 18, 50–5 (2009).
Simon, C., Sakkas, D., Gardner, D. K. & Critchley, H. O. D. Biomarkers in reproductive medicine: the quest for new answers. Hum. Reprod. Update 21, 695–697 (2015).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35, W193–200 (2007).
Reimand, J., Arak, T. & Vilo, J. G:Profiler - A web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res. 39, 1–9 (2011).
Paraskevopoulou, M. D. et al. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 41, W169–73 (2013).
Agarwal, V., Bell, G. W., Nam, J.-W. & Bartel, D. P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4 (2015).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–73 (2014).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 (2010).
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H. & Yang, J.-H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–7 (2014).
Krjutškov, K. et al. Single-cell transcriptome analysis of endometrial tissue. Hum. Reprod. 31, 844–53 (2016).
Raposo, G. & Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. 200, 373–383 (2013).
Kao, L. C. et al. Global gene profiling in human endometrium during the window of implantation. Endocrinology 143, 2119–2138 (2002).
Borthwick, J. M. et al. Determination of the transcript profile of human endometrium. Mol Hum Reprod 9, 19–33 (2003).
Carson, D. D. et al. Changes in gene expression during the early to mid-luteal (receptive phase) transition in human endometrium detected by high-density microarray screening. Mol Hum Reprod 8, 871–879 (2002).
Riesewijk, A. et al. Gene expression profiling of human endometrial receptivity on days LH+2 versus LH+7 by microarray technology. Mol Hum Reprod 9, 253–264 (2003).
Mirkin, S. et al. In search of candidate genes critically expressed in the human endometrium during the window of implantation. Hum Reprod 20, 2104–2117 (2005).
Julkunen, M. et al. Secretory endometrium synthesizes placental protein 14. Endocrinology 118, 1782–6 (1986).
Allegra, A. et al. Endometrial expression of selected genes in patients achieving pregnancy spontaneously or after ICSI and patients failing at least two ICSI cycles. Reprod. Biomed. Online 25, 481–91 (2012).
Apparao, K. B. et al. Osteopontin and its receptor alphavbeta(3) integrin are coexpressed in the human endometrium during the menstrual cycle but regulated differentially. J. Clin. Endocrinol. Metab. 86, 4991–5000 (2001).
Zhang, D., Lei, C. & Zhang, W. Up-regulated monoamine oxidase in the mouse uterus during the peri-implantation period. Arch. Gynecol. Obstet. 284, 861–866 (2011).
Macdonald, L. J. et al. Prokineticin 1 induces Dickkopf 1 expression and regulates cell proliferation and decidualization in the human endometrium. Mol. Hum. Reprod. 17, 626–36 (2011).
Serafini, P. et al. Protein profile of the luteal phase endometrium by tissue microarray assessment. Gynecol. Endocrinol. 25, 587–92 (2009).
Kitaya, K. et al. IL-15 expression at human endometrium and decidua. Biol. Reprod. 63, 683–7 (2000).
Nogawa Fonzar-Marana, R. R. et al. Expression of complement system regulatory molecules in the endometrium of normal ovulatory and hyperstimulated women correlate with menstrual cycle phase. Fertil. Steril. 86, 758–61 (2006).
Ohta, T. et al. Expression profiles of perforin, granzyme B and granulysin genes during the estrous cycle and gestation in the bovine endometrium. Anim. Sci. J. 85, 763–9 (2014).
Ugur, Y., Cakar, A. N., Beksac, M. S. & Dagdeviren, A. Activation antigens during the proliferative and secretory phases of endometrium and early-pregnancy decidua. Gynecol. Obstet. Invest. 62, 66–74 (2006).
Dolanbay, E. G. et al. Expression of trophinin and dipeptidyl peptidase IV in endometrial co-culture in the presence of an embryo: A comparative immunocytochemical study. Mol. Med. Rep. 13, 3961–8 (2016).
Ponnampalam, A. P. & Rogers, P. A. W. Cyclic changes and hormonal regulation of annexin IV mRNA and protein in human endometrium. Mol. Hum. Reprod. 12, 661–9 (2006).
Fujiwara, H. et al. Human endometrial epithelial cells express ephrin A1: possible interaction between human blastocysts and endometrium via Eph-ephrin system. J. Clin. Endocrinol. Metab. 87, 5801–7 (2002).
Germeyer, A. et al. Cell-type specific expression and regulation of apolipoprotein D and E in human endometrium. Eur. J. Obstet. Gynecol. Reprod. Biol. 170, 487–91 (2013).
Evans-Hoeker, E. et al. Endometrial BCL6 Overexpression in Eutopic Endometrium of Women With Endometriosis. Reprod. Sci. 23, 1234–41 (2016).
Yang, S. et al. Regulation of aromatase P450 expression in endometriotic and endometrial stromal cells by CCAAT/enhancer binding proteins (C/EBPs): decreased C/EBPbeta in endometriosis is associated with overexpression of aromatase. J. Clin. Endocrinol. Metab. 87, 2336–45 (2002).
Tajima, M., Harada, T., Ishikawa, T., Iwahara, Y. & Kubota, T. Augmentation of arginase II expression in the human endometrial epithelium in the secretory phase. J. Med. Dent. Sci. 59, 75–82 (2012).
Das, S., Vince, G. S., Lewis-Jones, I., Bates, M. D. & Gazvani, R. The expression of human alpha and beta defensin in the endometrium and their effect on implantation. J. Assist. Reprod. Genet. 24, 533–9 (2007).
Keator, C. S., Mah, K., Ohm, L. & Slayden, O. D. Estrogen and progesterone regulate expression of the endothelins in the rhesus macaque endometrium. Hum. Reprod. 26, 1715–28 (2011).
Rutanen, E.-M., Gonzalez, E., Said, J. & Braunstein, G. D. Immunohistochemical localization of the insulinlike growth factor binding protein-1 in female reproductive tissues by monoclonal antibodies. Endocr. Pathol. 2, 132–138 (1991).
Sedlmayr, P. et al. Localization of indoleamine 2,3-dioxygenase in human female reproductive organs and the placenta. Mol. Hum. Reprod. 8, 385–91 (2002).
Deng, L. et al. Expression and clinical significance of annexin A2 and human epididymis protein 4 in endometrial carcinoma. J. Exp. Clin. Cancer Res. 34, 96 (2015).
Altmäe, S., Kallak, T. K., Friden, B. & Stavreus-Evers, A. Variation in Hyaluronan-Binding Protein 2 (HABP2) Promoter Region is Associated With Unexplained Female Infertility. Reprod Sci 18, 485–492 (2011).
Mobasheri, A., Wray, S. & Marples, D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J. Mol. Histol. 36, 1–14 (2005).
Klimek, M. et al. Cycle dependent expression of endometrial metallothionein. Neuro Endocrinol. Lett. 26, 663–6 (2005).
Malette, B. et al. Large scale validation of human N-myc downstream-regulated gene (NDRG)-1 expression in endometrium during the menstrual cycle. Mol. Hum. Reprod. 9, 671–9 (2003).
Kottawatta, K. S. A. et al. MicroRNA-212 Regulates the Expression of Olfactomedin 1 and C-Terminal Binding Protein 1 in Human Endometrial Epithelial Cells to Enhance Spheroid Attachment In Vitro. Biol. Reprod. 93, 109 (2015).
Acknowledgements
This research was funded by a Marie Curie post-doctoral fellowship (FP7, no. 329812, NutriOmics); grant from University of Granada (Incorporación de jóvenes doctores); Estonian Ministry of Education and Research (grant IUT34-16); Enterprise Estonia (grant EU48695); the EU-FP7 Eurostars program (grant NOTED, EU41564); the EU-FP7 Marie Curie Industry-Academia Partnerships and Pathways (IAPP, grant SARM, EU324509); Horizon 2020 innovation programme (WIDENLIFE, 692065) and the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Author information
Authors and Affiliations
Contributions
Performed thorough literature search: S.A., M.S. Performed meta-analysis: U.V. Provided additional data for meta-analysis: L.A., P.G.L., K.G.-D., L.G., C.S. Performed additional statistical analyses: S.A., M.K., P.A., T.L.-P., V.K. Comprehensive validation experiments: M.K., M.S., T.L.-P., Merli S., A.V-M., K.K, A.S. Wrote the main body of the manuscript: S.A., M.K., U.V. Contributed to manuscript writing and editing: P.A., M.S., T.L.-P., V.K., Merli S., A.V.-M., K.K., L.A., P.G.L., K.G.-D., L.G., C.S., A.S. Authors have declared no competing interests.
Corresponding author
Ethics declarations
Competing Interests
Prof. Carlos Simón is the Chief Scientific Officer of Igenomiz, a Biotec Company that commercialize the ERA test. All the rest of the authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Altmäe, S., Koel, M., Võsa, U. et al. Meta-signature of human endometrial receptivity: a meta-analysis and validation study of transcriptomic biomarkers. Sci Rep 7, 10077 (2017). https://doi.org/10.1038/s41598-017-10098-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10098-3
- Springer Nature Limited
This article is cited by
-
Enhancing endometrial receptivity: the roles of human chorionic gonadotropin in autophagy and apoptosis regulation in endometrial stromal cells
Reproductive Biology and Endocrinology (2024)
-
MiR-124-3p negatively impacts embryo implantation via suppressing uterine receptivity formation and embryo development
Reproductive Biology and Endocrinology (2024)
-
Meta-analysis of endometrial transcriptome data reveals novel molecular targets for recurrent implantation failure
Journal of Assisted Reproduction and Genetics (2024)
-
Deciphering a shared transcriptomic regulation and the relative contribution of each regulator type through endometrial gene expression signatures
Reproductive Biology and Endocrinology (2023)
-
Can biomarkers identified from the uterine fluid transcriptome be used to establish a noninvasive endometrial receptivity prediction tool? A proof-of-concept study
Reproductive Biology and Endocrinology (2023)