

Abstract
Within population biobanks, incomplete measurement of certain traits limits the power for genetic discovery. Machine learning is increasingly used to impute the missing values from the available data. However, performing genome-wide association studies (GWAS) on imputed traits can introduce spurious associations, identifying genetic variants that are not associated with the original trait. Here we introduce a new method, synthetic surrogate (SynSurr) analysis, which makes GWAS on imputed phenotypes robust to imputation errors. Rather than replacing missing values, SynSurr jointly analyzes the original and imputed traits. We show that SynSurr estimates the same genetic effect as standard GWAS and improves power in proportion to the quality of the imputations. SynSurr requires a commonly made missing-at-random assumption but relaxes the requirements of existing imputation methods by not requiring correct model specification. We present extensive simulations and ablation analyses to validate SynSurr and apply it to empower the GWAS of dual-energy X-ray absorptiometry traits within the UK Biobank.






Similar content being viewed by others

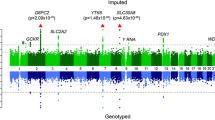
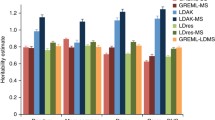
Data availability
This work used genotypes and phenotypes from the UKB. Summary statistics from the DEXA trait analysis will be deposited with the GWAS Catalog, and are available upon reasonable request in the interim.
Code availability
SurrogateRegression v.0.6.0.1 is available as an R54 package on the Comprehensive R Archive Network: https://CRAN.R-project.org/package=SurrogateRegression (ref. 56). The replication code for the analyses presented in this paper is available on GitHub at https://github.com/jianhuig/SyntheticSurrogateAnalysis (ref. 57).
References
Kurki, M. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Bycroft, C. et al. The UK Biobank resource with deep phenoty** and genomic data. Nature 562, 203–209 (2018).
Beesley, L. J. et al. The emerging landscape of health research based on biobanks linked to electronic health records: existing resources, statistical challenges, and potential opportunities. Stat. Med. 39, 773–800 (2020).
Tan, V. Y. & Timpson, N. J. The UK Biobank: a shining example of genome-wide association study science with the power to detect the murky complications of real-world epidemiology.Annu. Rev. Genomics Hum. Genet. 23, 569–589 (2022).
Wei, W.-Q. & Denny, J. C. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med. 7, 41 (2015).
Banda, J. M., Seneviratne, M., Hernandez-Boussard, T. & Shah, N. H. Advances in electronic phenoty**: from rule-based definitions to machine learning models. Annu. Rev. Biomed. Data Sci. 1, 53–68 (2018).
Allen, N., Sudlow, C., Peakman, T. & Collins, R. UK Biobank data: come and get it. Sci. Transl. Med. 6, 224ed4 (2014).
Littlejohns, T. J. et al. The UK Biobank imaging enhancement of 100,000 participants: rationale, data collection, management and future directions. Nat. Commun. 11, 2624 (2020).
Elliott, L. T. et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216 (2018).
Pirruccello, J. et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 11, 2254 (2020).
Alipanahi, B. et al. Large-scale machine-learning-based phenoty** significantly improves genomic discovery for optic nerve head morphology. Am. J. Hum. Genet. 108, 1217–1230 (2021).
Li, X. & Zhao, H. Automated feature extraction from population wearable device data identified novel loci associated with sleep and circadian rhythms. PLoS Genet. 16, e1009089 (2020).
Hormozdiari, F. et al. Imputing phenotypes for genome-wide association studies. Am. J. Hum. Genet. 99, 89–103 (2016).
Dahl, A. et al. A multiple-phenotype imputation method for genetic studies. Nat. Genet. 48, 466–472 (2016).
Zhang, Y. et al. High-throughput phenoty** with electronic medical record data using a common semi-supervised approach (PheCAP). Nat. Protoc. 14, 3426–3444 (2019).
Liao, K. P. et al. High-throughput multimodal automated phenoty** (MAP) with application to PheWAS. J. Am. Med. Inform. Assoc. 26, 1255–1262 (2019).
Cosentino, J. et al. Inference of chronic obstructive pulmonary disease with deep learning on raw spirograms identifies new genetic loci and improves risk models. Nat. Genet. 55, 787–795 (2023).
An, U. et al. Deep learning-based phenotype imputation on population-scale biobank data increases genetic discoveries. Nat. Genet. 55, 2269–2276 (2023).
Dahl, A. et al. Phenotype integration improves power and preserves specificity in biobank-based genetic studies of major depressive disorder. Nat. Genet. 55, 2082–2093 (2023).
Little, R. J. & Rubin, D. B. Statistical Analysis with Missing Data (John Wiley & Sons, 2002).
Wang, S., McCormick, T. H. & Leek, J. T. Methods for correcting inference based on outcomes predicted by machine learning. Proc. Natl Acad. Sci. USA 117, 30266–30275 (2020).
Hubbard, R. A., Tong, J., Duan, R. & Chen, Y. Reducing bias due to outcome misclassification for epidemiologic studies using EHR-derived probabilistic phenotypes. Epidemiology 31, 542–550 (2020).
Hong, C., Liao, K. P. & Cai, T. Semi-supervised validation of multiple surrogate outcomes with application to electronic medical records phenoty**. Biometrics 75, 78–89 (2019).
Rubin, D. Multiple Imputation for Nonresponse in Surveys (John Wiley & Sons, 1987).
Rubin, D. B. Multiple imputation after 18+ years. J. Am. Stat. Assoc. 91, 473–489 (1996).
van Buuren, S. Flexible Imputation of Missing Data (CRC, 2018).
Bartlett, J. W. & Hughes, R. A. Bootstrap inference for multiple imputation under uncongeniality and misspecification. Stat. Methods Med. Res. 29, 3533–3546 (2020).
Austin, P. C., White, I. R., Lee, D. S. & van Buuren, S. Missing data in clinical research: a tutorial on multiple imputation. Can. J. Cardiol. 37, 1322–1331 (2021).
Murray, J. S. Multiple imputation: a review of practical and theoretical findings. Stat. Sci. 33, 142–159 (2018).
McCaw, Z. R., Gaynor, S. M., Sun, R. & Lin, X. Leveraging a surrogate outcome to improve inference on a partially missing target outcome.Biometrics 79, 1472–1484 (2023).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Chen, T. & Guestrin, C. Xgboost: a scalable tree boosting system. in Proc. of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. https://dl.acm.org/doi/10.1145/2939672.2939785 (ACM, 2016).
Casella, B. & Berger, R. Statistical Inference (Duxbury/Thomson Learning, 2002).
Rubin, D. B. Inference and missing data. Biometrika 63, 581–592 (1976).
Body composition measurement protocol. BioBank https://biobank.ndph.ox.ac.uk/showcase/refer.cgi?id=1421 (2011).
DXA procedure within UKB imaging centre. BioBank https://biobank.ndph.ox.ac.uk/showcase/refer.cgi?id=502 (2015).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Weedon, M. et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 40, 575–583 (2008).
Liu, J. Z. et al. Genome-wide association study of height and body mass index in Australian twin families. Twin Res. Hum. Genet. 13, 179–193 (2010).
Meyre, D. et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 41, 157–159 (2009).
Willer, C. J. et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 41, 25–34 (2009).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 23, 120–133 (2022).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional map** and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
McCaw, Z., Lane, J., Saxena, R., Redline, S. & Lin, X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics 76, 1262–1272 (2020).
Robins, J. M. & Rotnitzky, A. Semiparametric efficiency in multivariate regression models with missing data. J. Am. Stat. Assoc. 90, 122–129 (1995).
Wang, X. & Wang, Q. Semiparametric linear transformation model with differential measurement error and validation sampling. J. Multivar. Anal. 141, 67–80 (2015).
Tong, J. et al. An augmented estimation procedure for EHR-based association studies accounting for differential misclassification. J. Am. Med. Inform. Assoc. 27, 244–253 (2020).
Po-Ru, L. et al. Efficient Bayesian mixed model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Seber, G. The Linear Model and Hypothesis (Springer, 2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Smith, G. D. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
McCaw, Z. SurrogateRegression: v0.6.0.1. Zenodo https://doi.org/10.5281/zenodo.10897842 (2024).
Gao, J. & Gronsbell, J. SyntheticSurrogateAnalysis: initial. Zenodo https://doi.org/10.5281/zenodo.10901237 (2024).
Acknowledgements
This work was supported by National Institutes of Health grant nos. R35-CA197449 and F31-HL140822 to Z.R.M.; nos. R35-CA197449, U19-CA203654, R01-HL163560, U01-HG012064 and U01-HG009088 to X.L.; and a Natural Sciences and Engineering Research Council of Canada grant no. RGPIN-2021-03734 and a Connaught New Researcher Award to J. Gronsbell. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.R.M., X.L. and J. Gronsbell designed the study and the experiments. Z.R.M. implemented the software with input from X.L. Z.R.M., J. Gao and J. Gronsbell performed the simulations. J. Gao conducted the analyses of the UKB data. Z.R.M. performed the overlap analysis. Z.R.M. and J. Gronsbell wrote the first draft of the manuscript; all coauthors provided intellectual revisions.
Corresponding authors
Ethics declarations
Competing interests
Z.R.M. is currently an employee of insitro, but he was not at the time of this work; his employer had no role in this study. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Robustness and precision of SynSurr with an uninformative and informative synthetic surrogate.
In all cases, the number of subjects with observed phenotypes was n = 103. The number of subjects with missing phenotypes was varied to achieve the indicated level of missingness. The standard estimator utilizes the observed values of Y only. In panel A, the synthetic surrogate has correlation ρ = 0.00 with the target phenotype, and is in fact independent of the target phenotype. Use of the SynSurr estimator with this uninformative surrogate results in no loss of efficiency relative to the standard analysis. In panel B, the synthetic surrogate has correlation ρ = 0.75 with the target phenotype. SynSurr becomes more efficient as the number of subjects with missing target outcomes increases.The center of the box plot is the median, the upper and lower bounds of the box are the 75th and 25th percentiles, and the whiskers extend from the minimum to the maximum. The number of simulation replicates is 5 × 103.
Extended Data Fig. 2 Signal recovery of SynSurr relative to the oracle GWAS for height and FEV1.
A slope of 1.0 (red line) indicates that the estimated effect sizes are consistent with the oracle effect sizes. Note that although the slope deviates from 1.0 at 90% missigness, the slope approaches 1.0 as missingness declines. The following figure, which assesses signal recovery for standard GWAS, provides a point of comparison for the R2 values.
Extended Data Fig. 3 Signal recovery of imputation-based approaches and SynSurr relative to the oracle GWAS for height with 50% missingness.
A slope of 1.0 (red line) indicates that the estimated effect sizes are consistent with the oracle effect sizes, whereas a slope deviating from 1.0 suggests the presence of bias. The estimated slope of the data is shown by the blue line. In (a) the surrogates and imputations were generated from a random forest model, whereas in (b) they were generated from a linear regression. In (c), the surrogates and imputations were permuted such that they were uncorrelated with the target outcome. In (d), the surrogates and imputations were negated such that the correlation changed direction but not magnitude.
Extended Data Fig. 4 Predicted vs. observed values of body composition phenotypes within the model-building and GWAS data sets.
A random forest was trained to predict each of the 6 body composition phenotypes, obtained via DEXA scan, using 4,584 subjects allocated to the model-building data set. The GWAS dataset consists of 29,577 unrelated subjects with body compositions measured via DEXA. Model inputs included age, sex, height, body weight, body mass index, and 5 impedance measures (whole body, left arm, right arm, left leg and right leg). The estimated slope of the data is shown by the blue line.
Extended Data Fig. 5 Distribution of predicted body masses comparing subjects with and without DEXA measurements.
The violin plot shows the kernel density estimation of the distribution of the data, with the tips of the violin indicating the maximum and minimum observed values among subjects. Sample sizes: n = 29, 577 independent subjects with DEXA measurements; n = 317, 921 subjects without DEXA measurements.
Extended Data Fig. 6 SynSurr remains unbiased and properly controls the type I error when the same data are utilized for model training and for GWAS.
The number of subjects with observed phenotypes was n = 103, while the number with missing phenotypes was varied to achieve the indicated level of missingness. The model that generated the synthetic surrogate was either trained in the GWAS data set or in an independent data set of size n = 103. Upper shows the distribution of effect sizes across 20 × 103. The true genetic effect size is βG = 0.1. The center of the box plot is the median, the upper and lower bounds of the box are the 75th and 25th percentiles, and the whiskers extend from the 5th to the 95th percentile. Lower shows the average χ2 statistic under H0: βG = 0 across 50 × 103 simulation replicates, for which the expected value is 1.0. Error bars are 95% confidence intervals for the mean. Panel A (left) considers a ‘misspecified’ (k = 2) model that can only capture quadratic dependence of Y on X, while Panel B (right) considers a ‘correctly specified’ model (k = 3) that can capture the cubic dependence. As seen, the validity of SynSurr is not contingent on correct specification of the surrogate model.
Extended Data Fig. 7 Survey of assumptions surrounding missing phenotypic data in GWAS.
The methods sections of all studies contributing summary statistics to the GWAS catalog between May 1st and November 1st, 2023, were manually reviewed. Among 47 studies, 24 did not address missing phenotypic data. Of the 23 remaining, 21 made an assumption of missing at random (MAR) or missing completely at random (MCAR).
Supplementary information
Supplementary Information
Supplementary Figs. 1–22, Tables 1–22, Methods, Simulations, Genotype quality control and Survey of missing data in the GWAS.
Supplementary Data 1
Accession numbers for the GWAS Catalog overlap analysis.
Supplementary Data 2
DEXA body composition phenotype gene set enrichment analysis.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
McCaw, Z.R., Gao, J., Lin, X. et al. Synthetic surrogates improve power for genome-wide association studies of partially missing phenotypes in population biobanks. Nat Genet (2024). https://doi.org/10.1038/s41588-024-01793-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41588-024-01793-9
- Springer Nature America, Inc.