
Abstract
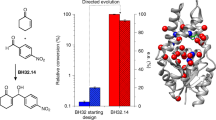
New enzyme catalysts are usually engineered by repurposing the active sites of natural proteins. Here we show that design and directed evolution can be used to transform a non-natural, functionally naive zinc-binding protein into a highly active catalyst for an abiological hetero-Diels–Alder reaction. The artificial metalloenzyme achieves >104 turnovers per active site, exerts absolute control over reaction pathway and product stereochemistry, and displays a catalytic proficiency (1/KTS = 2.9 × 1010 M−1) that exceeds all previously characterized Diels–Alderases. These properties capitalize on effective Lewis acid catalysis, a chemical strategy for accelerating Diels–Alder reactions common in the laboratory but so far unknown in nature. Extension of this approach to other metal ions and other de novo scaffolds may propel the design field in exciting new directions.





Similar content being viewed by others


Data availability
Structural data obtained by X-ray crystallography were deposited in the Protein Data Bank (PDB) or the Cambridge Crystallographic Data Centre (CCDC), and are available with the following accession codes: DA7W16S (7BWW), DA7W16G,K58Q,L77R,T78R (6YPI), racemic exo-3 (1972193), 4R,6R-endo-4 (1972197) and R-(+)-phenylethylamine phosphate (1972198). All relevant data are provided in the figures, Extended Data and Supplementary Information; alternatively, the data are available from the corresponding author upon request.
References
Nicolaou, K. C., Snyder, S. A., Montagnon, T. & Vassilikogiannakis, G. The Diels–Alder reaction in total synthesis. Angew. Chem. Int. Ed. 41, 1668–1698 (2002).
Stocking, E. M. & Williams, R. M. Chemistry and biology of biosynthetic Diels–Alder reactions. Angew. Chem. Int. Ed. 42, 3078–3115 (2003).
Minami, A. & Oikawa, H. Recent advances of Diels–Alderases involved in natural product biosynthesis. J. Antibiot. 69, 500–506 (2016).
Jeon, B.-S., Wang, S.-A., Ruszczycky, M. W. & Liu, H.-W. Natural [4 + 2]-cyclases. Chem. Rev. 117, 5367–5388 (2016).
Jamieson, C. S., Ohashi, M., Liu, F., Tang, Y. & Houk, K. N. The expanding world of biosynthetic pericyclases: cooperation of experiment and theory for discovery. Nat. Prod. Rep. 36, 698–713 (2019).
Romesberg, F. E., Spiller, B., Schultz, P. G. & Stevens, R. C. Immunological origins of binding and catalysis in a Diels-Alderase antibody. Science 279, 1929–1933 (1998).
Xu, J. et al. Evolution of shape complementarity and catalytic efficiency from a primordial antibody template. Science 286, 2345–2348 (1999).
Gouverneur, V. E. et al. Control of the exo and endo pathways of the Diels-Alder reaction by antibody catalysis. Science 262, 204–208 (1993).
Siegel, J. B. et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010).
Preiswerk, N. et al. Impact of scaffold rigidity on the design and evolution of an artificial Diels-Alderase. Proc. Natl Acad. Sci. USA 111, 8013–8018 (2014).
Pindur, U., Lutz, G. & Otto, C. Acceleration and selectivity enhancement of Diels–Alder reactions by special and catalytic methods. Chem. Rev. 93, 741–761 (1993).
Fringuelli, F., Piermatti, O., Pizzo, F. & Vaccaro, L. Recent advances in Lewis acid catalyzed Diels–Alder reactions in aqueous media. Eur. J. Org. Chem. 2001, 439–455 (2001).
Reetz, M. T. & Jiao, N. Copper–phthalocyanine conjugates of serum albumins as enantioselective catalysts in Diels–Alder reactions. Angew. Chem. Int. Ed. 45, 2416–2419 (2006).
Podtentenieff, J., Taglieber, A., Bill, E., Reijerse, E. J. & Reetz, M. T. An artificial metalloenzyme: creation of a designed copper binding site in a thermostable protein. Angew. Chem. Int. Ed. 49, 5151–5155 (2010).
Bos, J., Fusetti, F., Driessen, A. J. M. & Roelfes, G. Enantioselective artificial metalloenzymes by creation of a novel active site at the protein dimer interface. Angew. Chem. Int. Ed. 51, 7472–7475 (2012).
Ghattas, W. et al. Receptor-based artificial metalloenzymes on living human cells. J. Am. Chem. Soc. 140, 8756–8762 (2018).
Studer, S. et al. Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 362, 1285–1288 (2018).
Der, B. S. et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc. 134, 375–385 (2012).
Ohashi, M. et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017).
Richter, F., Leaver-Fay, A., Khare, S. D., Bjelic, S. & Baker, D. De novo enzyme design using Rosetta3. PLoS ONE 6, e19230 (2011).
Lei, G. et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 12, 620–628 (2020).
Radzicka, A. & Wolfenden, R. A proficient enzyme. Science 267, 90–93 (1995).
Kim, H. J., Ruszczycky, M. W., Choi, S.-H., Liu, Y.-N. & Liu, H.-W. Enzyme-catalysed [4+2] cycloaddition is a key step in the biosynthesis of spinosyn A. Nature 473, 109–112 (2011).
Fage, C. D. et al. The structure of SpnF, a standalone enzyme that catalyses [4 + 2] cycloaddition. Nat. Chem. Biol. 11, 256–258 (2015).
Byrne, M. J. et al. The catalytic mechanism of a natural Diels–Alderase revealed in molecular detail. J. Am. Chem. Soc. 138, 6095–6098 (2016).
Zhang, Z. et al. Enzyme-catalyzed inverse-electron demand Diels–Alder reaction in the biosynthesis of antifungal ilicicolin H. J. Am. Chem. Soc. 141, 5659–5663 (2019).
Kiss, G., Çelebi-Ölçüm, N., Moretti, R., Baker, D. & Houk, K. N. Computational enzyme design. Angew. Chem. Int. Ed. 52, 5700–5752 (2013).
Schwizer, F. et al. Artificial metalloenzymes: reaction scope and optimization strategies. Chem. Rev. 118, 142–231 (2018).
Zeymer, C. & Hilvert, D. Directed evolution of protein catalysts. Annu. Rev. Biochem. 87, 131–157 (2018).
Yamamoto, H. (ed.) Lewis Acids in Organic Synthesis (Wiley-VCH, 2000).
Huang, P.-S., Boyken, S. E. & Baker, D. The coming of age of de novo protein design. Nature 537, 320–327 (2016).
Acknowledgements
We thank M. Solar and N. Trapp from the Small Molecule Crystallography Center at ETH Zürich for structure elucidation. We also thank B. Blattmann from the Protein Crystallization Center at the University of Zürich, the teams from the Mass Spectrometry service and the NMR facility at the Laboratory for Organic Chemistry at ETH Zürich and the staff at the Swiss Light Source at the Paul Scherrer Institute for technical support. This work was supported by the Swiss National Science Foundation (SNSF) and ETH Zürich. G.J.-O. acknowledges grants from the Spanish Ministry of Science and Innovation (MCI) co-financed with FEDER funds (RTI2018-099592-B-C22 and RYC-2013-14706) and the Mizutani Foundation for Glycoscience (200077).
Author information
Authors and Affiliations
Contributions
D.H., G.J.-O. and K.N.H. conceived the project. G.J.-O. and K.N.H. carried out the computational enzyme design. H.A.B. carried out preliminary experiments. S.S. designed the experimental strategy. S.S. and A.C. selected and tested the initial constructs. S.B. and S.S. evolved and characterized the enzyme and analysed the data. S.B., Y.O. and R.C.H. synthesized the substrates. S.B. performed the reaction characterization, protein and small molecule crystallization. T.M. crystallized the enzyme and solved its structure. Y.Z. and G.J.-O. performed computational MD simulations and DFT calculations. D.H. supervised the research. D.H., S.B. and K.N.H. wrote the paper with contributions from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 DFT calculations for competing Diels-Alder and hetero-Diels-Alder pathways.
a, Diels-Alder and hetero-Diels-Alder reactions of the azachalcone 1 and 3-vinylindole 2 in SMD water [calculated at the ωB97X-D/6-311 + +g(2d,p)/SMD(water)//ωB97X-D/6-311 G(d,p)/SMD (water) level]. TS1-endo is ambimodal and affords both 3-endo and 4-endo. Experimentally, only 3-exo and 4-endo are observed. b, The endo hetero-Diels-Alder reaction of 1 and 2 in the presence of [Zn(OH)(H2O)2]+, calculated at the ωB97X-D/6-311 + +G(2d,p)/SMD(water)//ωB97X-D/6-311 G(d,p)/ SMD(water) level (the LANL2DZ and SDD effective core potentials were used for Zn in geometry optimisation and single energy calculations, respectively), occurs via a stepwise mechanism that favours formation of 4-endo. Relative enthalpies (H) and free energies (G) are shown in blue and red, and are reported in kcal/mol. Labelled distances are in ångströms.
Extended Data Fig. 2 Chemo- and stereoselectivity.
a, HPLC chromatogram (at 350 nm) and crystal structure (Supplementary Table 11) of the racemic exo Diels-Alder product 3 (only one enantiomer is shown for clarity). b, HPLC chromatogram of the racemic hetero-Diels-Alder products 4 and crystal structure of the enzymatically synthesised (4 R,6 R)-endo hetero-Diels-Alder product. The hydrogen atoms on the central six-membered ring are shown for the X-ray crystal structures in a and b. c, HPLC analysis of the Diels-Alder (3.36 min) and hetero-Diels-Alder (3.06 min) product profile under different conditions. d, Chiral HPLC chromatograms of the hetero-Diels-Alder reaction products from reactions without protein (grey) and reactions catalysed by DA0 (pink) and DA7 (orange). Retention times vary due to different column temperatures (40 °C vs room temperature).
Extended Data Fig. 3 Directed evolution of DA7.
a, Simplified schematic showing the steps of computational design and directed evolution leading to DA7. b, Amino acid alignment of evolutionary parent DA0, evolved variant DA7, and two intermediate variants, DA1 and DA4. Mutations distinguishing the variants are highlighted in orange. The two mutations suggested by computation are shown in pink.
Extended Data Fig. 4 Kinetic characterisation of buffer-catalysed reaction, intermediate DA variants, and DA7.
a, Non-enzymatic background reaction between Diels-Alder substrates 1 and 2 in buffer. The rates were determined in 20 mM MOPS, pH 8, 3.5% DMSO, 10 µM Zn(II), 1 mg/mL BSA at 25 °C. Error bars denote s.d. b–d, Michaelis-Menten plots for the reaction between 1 and 2 catalysed by the parental scaffold DA0 and evolutionary intermediates DA1 and DA4. An analogous plot for DA7 is shown in Fig. 2c in the main text. For DA0 (b), error bars indicate the standard deviation between three biological replicates. e, Relative activities of DA7 and the variants DA7W16S, DA7C35H and DA7Q80A, which were respectively prepared to aid crystallisation and probe mechanism. Mutation of the metal binding site (DA7C35A/H61A/H65A) or removal of zinc with EDTA leads to complete loss of activity. kobs values were determined using 30 µM 1 and 60 µM 2, where [1] and [2] « KM,1 and KM,2 and thus (v0/[E]0)/([1]0[2]0) ≈ kcat/(KM,1KM,2). f, Total turnovers for DA7 were determined using 200 µM 1 and 400 µM 2 and 10 nM enzyme. The consumption of azachalcone was corrected for contributions from the background reaction.
Extended Data Fig. 5 Change in metal binding site and crossing angle.
a, b, Comparison of the metal binding sites of MID1 (a) and DA7 (b). MID1 coordinates the Zn(II) ion (yellow sphere) with three histidine residues (grey sticks). A fourth non-coordinating histidine points away from the metal binding site. H35C and H39V mutations (orange sticks) altered the coordination sphere of the catalytic Zn(II) ion during evolution. c, Overlay of parental MID1 (grey) and DA7 (orange) shows the decrease in crossing angle of the two helix-turn-helix fragments by > 30°. d, Calculated acute crossover angles θ between helices H1–H4 of MID1 and DA7W16S.
Extended Data Fig. 6 Discrimination of enantiomeric transition states by DA7.
a, Whereas the equilibrated rate-limiting transition state leading to the experimentally observed (4R, 6R)-hetero-Diels-Alder product binds productively at the DA7 active site (TS1-endo, green; see also Fig. 3 in the main text), severe steric clashes between the indole moiety of the enantiomeric TS1-endo (yellow sticks) and the protein backbone are observed if the azachalcone moiety chelates the catalytic zinc ion. b, Restrained molecular dynamics simulations show that the clashes between the enantiomeric TS1-endo and DA7 are only relieved at the expense of zinc coordination, explaining why the evolved enzyme does not produce the (4S, 6S)-hetero-Diels-Alder product. Labelled distances are in angstrom. c, d, Surface representation of DA7 showing the binding pocket with the preferred transition state docked at the active site in stick (c) and space-filling (d) representation.
Extended Data Fig. 7 A catalytically relevant hydrogen bond network in DA7.
Molecular dynamics simulations of INT1-endo bound to DA7 show fluctuations in hydrogen bond distances between a, the 3-vinylindole moiety of the ligand and Gln80, b, Gln80 and Gln31, and c, Gln31 and Tyr84. Gln80 maintains hydrogen bonding interactions with both the ligand and Gln31 throughout the simulation, whereas Gln31 and Tyr84 interact for fraction of the time.
Supplementary information
Supplementary Information
Supplementary materials and methods, Tables 1–11, Data 1–16 and refs. 1–78.
Supplementary Data 1
Crystallographic information file for racemic exo-3 (CCDC 1972193).
Supplementary Data 2
Crystallographic information file for 4R,6R-endo-4 (CCDC 1972197).
Supplementary Data 3
Crystallographic information file for R-(+)-phenylethylamine phosphate (CCDC 1972198).
Supplementary Data 4
XYZ coordinates from DFT calculations.
Rights and permissions
About this article

Cite this article
Basler, S., Studer, S., Zou, Y. et al. Efficient Lewis acid catalysis of an abiological reaction in a de novo protein scaffold. Nat. Chem. 13, 231–235 (2021). https://doi.org/10.1038/s41557-020-00628-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-020-00628-4
- Springer Nature Limited