
Abstract
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease and arises from dopamine (DA) neuron death selectively in the substantia nigra pars compacta (SNc). Rit2 is a reported PD risk allele, and recent single cell transcriptomic studies identified a major RIT2 cluster in PD DA neurons, potentially linking Rit2 expression loss to a PD patient cohort. However, it is still unknown whether Rit2 loss itself impacts DA neuron function and/or viability. Here we report that conditional Rit2 silencing in mouse DA neurons drove motor dysfunction that occurred earlier in males than females and was rescued at early stages by either inhibiting the DA transporter (DAT) or with L-DOPA treatment. Motor dysfunction was accompanied by decreased DA release, striatal DA content, phenotypic DAergic markers, DA neurons, and DAergic terminals, with increased pSer129-alpha synuclein and pSer935-LRRK2 expression. These results provide clear evidence that Rit2 loss is causal for SNc cell death and motor dysfunction, and reveal key sex-specific differences in the response to Rit2 loss.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a complex, progressive, neurodegenerative disorder characterized by SNc DA neuron (DAN) death1,2. PD prevalence is higher in males and PD symptoms often go unnoticed until >75% of SNc neurons have died3,4. Phenotypically, PD patients exhibit profound motor impairment that includes bradykinesia, resting tremor, muscular rigidity, lack of coordination, and postural instability5. These symptoms are due to SNc DAN cell death and concomitant diminished striatal DA signaling, and PD therapeutic strategies typically aim to boost DA production in the remaining DAN population6.
Rit2 (AKA: Rin, Ras-like in neurons) is a small, neuronal, ras-like GTPase with enriched expression in SNc DANs7. Rit2 directly interacts with the DA transporter (DAT), and is required for regulated DAT membrane trafficking8,9,10. In cell culture models, Rit2 is required for EGF- and NGF-mediated neurite outgrowth, NGF-mediated ERK phosphorylation, and cell viability11,12,13,14. Genome-wide association studies (GWAS) link Rit2 genetic anomalies to PD15,16,17,18,19,20,21,57. Gait testing apparatus consisted of a 10 cm x 36 cm runway with 14 cm high foamboard walls and a dark box at the opposing end. Fresh, legal-size paper was placed on the benchtop under the runway for each trial. Mouse forepaws and hind-paws were dipped in non-toxic orange and blue tempera paint, respectively, and mice were placed on the paper at the open end of the runway and allowed to traverse to the closed box at the opposing end. Three trials were performed per mouse and stride length, stride width and tow spread were measured for both fore- and hindlimbs. Number of completed trials was also quantified. Experimenters and data analysts were double-blinded to mouse IDs.
Grip strength
Four-limb grip strength was measured using the Bioseb Grip Strength Test (BIO-GS3) equipped with mesh grip grid for mice. Mice were suspended by tail over the mesh and lowered onto it until all 4 paws grasped the mesh. The mouse was then pulled backwards gently on the horizontal plane until it released from the mesh. The maximal force applied was recorded for 3 consecutive trials and averaged.
Tissue harvesting and immunoblotting
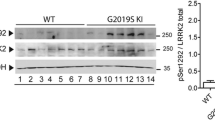
Striata were collected by preparing 300 µm coronal sections on a Vibratome as previously described8,9. Sections were collected through the entire striatum, dorsal and ventral striata were sub-dissected, and slices encompassing each region were pooled for each independent mouse. Tissue was lysed in RIPA buffer (10 mM Tris, pH 7.4; 150 mM NaCl; 1.0 mM EDTA; 0.1% SDS, 1% Triton X-100, 1% Na deoxycholate) supplemented with protease inhibitors (1.0 mM phenylmethylsulfonyl fluoride and 1.0 g/mL each leupeptin, aprotinin, and pepstatin) and Phosphatase inhibitor cocktail V (EMD Millipore). Mechanical tissue disruption was also performed by triturating sequentially through a 200 µL pipette tip, 22-, and 26- gauge tech tips and solubilized by rotating (30 min 4°C). Insoluble material was removed by centrifugation (15 min, 18 K x g, 4°C). Lysate protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific). Protein samples were denatured in an equal volume of 2x Laemmli sample buffer and were either rotated (30 min, RT) for membrane protein immunoblots or boiled (5 min) for soluble protein immunoblots. Proteins were resolved by SDS-Page, transferred to nitrocellulose membranes, and the indicated proteins were detected and quantified by immunoblotting with the following antibodies: rat anti-DAT (MAB369, Millipore; 1:2000), rabbit anti-TH (AB152, Millipore, 1:10000), rabbit anti-pSer40 TH (AB5935, Millipore, 1:5000), rabbit anti-αSyn, rabbit anti-pSer129-αSyn, anti-LRRK2, anti-pSer935 LRRK2, mouse anti-actin (Santa Cruz, 1:5000). Secondary antibodies conjugated to horseradish peroxidase were all from Jackson ImmunoResearch and immunoreactive bands were visualized by chemiluminescence using SuperSignal West Dura (Thermo Scientific). Immunoblotting solutions were prepared in either PBS-T, or TBS-T (137 mM NaCl, 2.7 mM KCl, 19 mM Tris base, ph7.4, 0.1% Tween20) when probing for phosphoproteins. Non-saturating immunoreactive bands were detected using either VersaDoc 5000MP or Chemidoc imaging stations (Bio-Rad) and were quantified using Quantity One software (Bio-Rad). Representative blots shown for a given condition were cropped from the same exposure of the same immunoblot and spliced together for presentation purposes only. Splice margins are indicated with a line. All compared blots were processed in parallel and derive from the same experiments.
Fast-scan cyclic voltammetry
Mice were sacrificed by cervical dislocation and rapid decapitation. Heads were immediately submerged in ice-cold NMDG cutting solution, pH 7.3-7.4 (20 mM HEPES, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 25 mM glucose, 0.5 mM CaCl2·4H2O. 10 mM MgSO4·7H2O, 92mM N-methyl-D-glucamine, 2 mM thiourea, 5 mM Na+-ascorbate, 3 mM Na+-pyruvate). Brains were removed, glued to the stage of a VT1200S Vibroslicer (Leica) and submerged in ice-cold, oxygenated cutting solution. 300 µm slices were prepared and were hemisected along the midline prior to recovering in ACSF (125 mM NaCl, 2.5 mM KCl, 1.24 mM NaH2PO4, 26 mM NaHCO3, 11 mM glucose, 2.4 mM CaCl2·4H2O,1.2 mM MgCl2·6H2O, pH 7.4) at 31°C for a minimum of 1 hour prior to recording. Hemislices were moved to the recording chamber and were perfused with oxygenated ASCF supplemented with 500 µM Na-Ascorbate. Glass pipettes containing a 7 µm carbon-fiber microelectrode were prepared and preconditioned in ASCF by applying triangular voltage ramps ( − 0.4 to +1.2 and back to −0.4 V at 400 V/s), delivered at 60 Hz for 1 hour. Recordings were performed at 10 Hz. Electrodes were calibrated to a 1 µM DA standard prior to recording. Electrodes were positioned in DS and DA transients were electrically evoked with a 250 µA rectangular pulse every 2 min, using a concentric bipolar electrode placed ~100 µm from the carbon fiber electrode. Data were collected with a 3-electrode headstage, using an EPC10 amplifier (Heka) after low-pass filter at 10 kHz and digitized at 100 kHz, using Patchmaster software (Heka). A stable baseline was achieved after evoking six consecutive DA transients, after which experimental data were collected. Each biological replicate is the average of three evoked DA transients/slice, and a minimum of 3 independent mice were used to gather data from the indicated number of slices in each experiment. Data were analyzed in Igor Pro, using the Wavemetrics FSCV plugin (gift of Veronica Alvarez, NIAAA). Peak amplitudes were measured for each individual DA transient, and tau was calculated as 1/e according to the equation: y = y0 + A((x-x0)/tau)).
Mass spectrometry
Sample preparation
Brains were harvested, 1.0 mm coronal sections were prepared and bilateral 1.0mm2 punches were each taken from dorsal and ventral striata. Each bilateral pair was solubilized in 10 µL internal standard solution (200 µM 13C4-GABA and 1 µM 2H3-DA in water with 500 µM ascorbic acid and 0.1% formic acid) and 50 µl ice-cold acetonitrile with 1% formic acid. Samples were vortexed twice for 0.5 min with a 1 min incubation on ice between vortexing and were sonicated in an ice-water bath until tissue was completely disrupted. Samples were centrifuged (10 min, 16,000 x g) and supernatants were collected for LC/MS/MS analysis. A standard (STD) solution containing 200 µM GABA, 1 µM dopamine, 500 µM ascorbic acid and 0.1% formic acid was also prepared.
LC/MS/MS
10 µl samples were injected in triplicate into a Thermo Scientific Ultimate 3000 HPLC system on a SeQuant ZIC-cHILIC column (2.1 ×100 mm, 3 µm) with a ZIC-cHILIC guard column (2.1 ×20 mm, 5 µm), coupled with a Thermo Scientific TSQ Quantiva triple quadrupole mass spectrometer. The mobile phase was water with 0.1% formic acid (A) and acetonitrile (B), and the elution program was as follows: 0 min 25% A, 0.5 min 25% A, 4.5 min 45% A, 5.0 min 70% A, 8.0 min 70% A, 8.1 min 25% A, 12.0 min 25% A at 0.2 mL/min. Ionization was operated in the positive mode with the voltage of 4.2 kV. The parameters were set as follow: sheath gas, 35 Arb, aux gas, 15 Arb, vaporizer temperature, 250 °C, ion transfer tube temperature, 325 °C. Multiple reaction monitoring (MRM) was performed using a cycle time of 0.3 s, CID gas pressure of 1.5 mTorr, Q1 resolution (FWHM) of 0.7 and Q3 resolution (FWHM) of 0.7. The MRM transitions 104.1 > 87 (GABA), 108.1 > 91 (13C4-GABA), 154.1 > 91 (dopamine) and 157.1 > 93 (2H3-dopamine) were selected for quantification. All data was integrated and processed in Xcalibur (Version 2.2, Thermo Scientific).
Stereological analysis and Immunohistochemistry/confocal microscopy
Briefly, mice were perfused and fixed with freshly made 4% paraformaldehyde (PFA) in PBS. Brains were removed immediately and fixed again in 4% PFA followed by equilibration in 30% sucrose in PBS. Midbrains were removed for stereological analysis, and forebrains were used for immunohistochemistry/confocal microscopy.
Stereological analysis
SNc total and TH+ neurons were quantified as previously described58. Fixed brains were imbedded in the OCT-compound media (Sakura) and frozen in liquid nitrogen. 40 µm cryosections were prepared through the midbrain a Leica CM3050s cryostat, and were stored in an antifreeze media containing 30% ethylene glycol, 25% glycerol, and 5% phosphate buffer. For stereology counting, 1 in every 5 sections was selected with a random start and a total of 6 brain slices on average were used for each mouse for IHC labeling for TH, including DAB enhancement, followed by Cresyl violet staining to reveal all neurons. Substantia nigra pars compacta was imaged using a Zeiss Axioplan 2 microscope equipped with a 20X objective, and Stereo Investigator was used to estimate the total number of neurons in the region of interest using the following parameters: frame sizes, 150 ×150 µm; grid sizes, 250 ×250 µm; top guard zone height, 2 µm; and optical dissector height, 8 µm. These parameters yielded a coefficient of error <10% throughout the analysis. Total cell numbers measured were weighted to section thickness for each mouse and were averaged across each cohort. Investigators performing stereological counting were blinded to mouse identity.
Immunohistochemistry/confocal microscopy
25 µm coronal sections through the striatum were prepared using a microtome and were co-stained with rabbit anti-pSer129-Syn (Cell Signaling #23706; 1:500) and chicken anti-TH (Millipore #AB9702, 1:500), followed by staining with secondary antibodies preabsorbed against mouse (Alexa568-goat anti-rabbit, Alexa647-donkey anti-chicken, Jackson ImmunoResearch). Z-stacks in the dorsal striatum were acquired with a Zeiss 700 LSM scanning confocal microscope using 555 nm and 647 nm lasers, and were pseudocolored to red and green, respectively. Acquisition settings within each channel (pinhole size, digital gain, and laser strength) were identical across all samples. Z-stacks were imported into ImageJ software where a representative plane was chosen, channels were separated (for individual red and green images), and images exported as tiff files. Files were subsequently imported into Adobe Photoshop, and levels were adjusted identically across all images.
Statistics
Data analysis was performed with GraphPad Prism software. All data were assessed for normality and nonparametric tests were applied if data distribution was non-Gaussian. Outliers in each data set were identified using either Grubb’s or Rout’s outlier tests, with a or Q values set at 0.05 or 5%, respectively, and were removed from further analysis. Significant differences between two values were determined using either a one-tailed, two-tailed, or paired Student’s t test, as indicated. Differences amongst more than two conditions were determined using one-way or two-way ANOVA, as appropriate, and significant differences among individual values within the group were determined by post-hoc multiple comparison tests, as described for each experiment.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Hartmann, A. Postmortem studies in Parkinson’s disease. Dialog. Clin. Neurosci. 6, 281–293 (2004).
Dickson, D. W. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2, a009258 (2012).
Pringsheim, T., Jette, N., Frolkis, A. & Steeves, T. D. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 29, 1583–1590 (2014).
Lees, A. J. Unresolved issues relating to the shaking palsy on the celebration of James Parkinson’s 250th birthday. Mov. Disord. 22, S327–334 (2007).
Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 (2008).
Tambasco, N., Romoli, M. & Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 16, 1239–1252 (2018).
Zhou, Q., Li, J., Wang, H., Yin, Y. & Zhou, J. Identification of nigral dopaminergic neuron-enriched genes in adult rats. Neurobiol. Aging. 32, 313–326 (2011).
Fagan, R. R. et al. Dopamine transporter trafficking and Rit2 GTPase: Mechanism of action and in vivo impact. J. Biol. Chem. 295, 5229–5244 (2020).
Kearney, P. J. et al. Presynaptic Gq-coupled receptors drive biphasic dopamine transporter trafficking that modulates dopamine clearance and motor function. J. Biol. Chem. 299, 102900 (2023).
Navaroli, D. M. et al. The plasma membrane-associated GTPase Rin interacts with the dopamine transporter and is required for protein kinase C-regulated dopamine transporter trafficking. J. Neurosci. 31, 13758–13770 (2011).
Hoshino, M. & Nakamura, S. The Ras-like small GTP-binding protein Rin is activated by growth factor stimulation. Biochem. Biophys. Res. Commun. 295, 651–656 (2002).
Spencer, M. L., Shao, H., Tucker, H. M. & Andres, D. A. Nerve growth factor-dependent activation of the small GTPase Rin. J Biol. Chem. 277, 17605–17615 (2002).
Hoshino, M. & Nakamura, S. Small GTPase Rin induces neurite outgrowth through Rac/Cdc42 and calmodulin in PC12 cells. J Cell Biol. 163, 1067–1076 (2003).
Shi, G. X., Han, J. & Andres, D. A. Rin GTPase couples nerve growth factor signaling to p38 and b-Raf/ERK pathways to promote neuronal differentiation. J. Biol. Chem. 280, 37599–37609 (2005).
Latourelle, J. C., Dumitriu, A., Hadzi, T. C., Beach, T. G. & Myers, R. H. Evaluation of Parkinson disease risk variants as expression-QTLs. PLoS One 7, e46199 (2012).
Pankratz, N. et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann. Neurol. 71, 370–384 (2012).
Emamalizadeh, B. et al. RIT2, a susceptibility gene for Parkinson’s disease in Iranian population. Neurobiol. Aging. 35, e27–e28 (2014).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014).
Lu, Y. et al. Genetic association of RIT2 rs12456492 polymorphism and Parkinson’s disease susceptibility in Asian populations: a meta-analysis. Sci. Rep. 5, 13805 (2015).
Liu, Z. H. et al. Assessment of RIT2 rs12456492 association with Parkinson’s disease in Mainland China. Neurobiol. Aging 36, 1600 e1609–1611 (2015).
Wang, J. Y. et al. The RIT2 and STX1B polymorphisms are associated with Parkinson’s disease. Parkinson. Relat. Disord. 21, 300–302 (2015).
Zhang, X., Niu, M., Li, H. & **e, A. RIT2 rs12456492 polymorphism and the risk of Parkinson’s disease: A meta-analysis. Neurosci Lett 602, 167–171 (2015).
Chan, G. et al. Trans-pQTL study identifies immune crosstalk between Parkinson and Alzheimer loci. Neurol. Genet. 2, e90 (2016).
Emamalizadeh, B. et al. RIT2 Polymorphisms: Is There a Differential Association? Mol. Neurobiol. 54, 2234–2240 (2017).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516 (2017).
Daneshmandpour, Y., Darvish, H. & Emamalizadeh, B. RIT2: responsible and susceptible gene for neurological and psychiatric disorders. Mol. Genet. Genom. 293, 785–792 (2018).
Glessner, J. T. et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA 107, 10584–10589 (2010).
Liu, H., Talalay, P. & Fahey, J. W. Biomarker-Guided Strategy for Treatment of Autism Spectrum Disorder (ASD). CNS Neurol. Disord. Drug Targets 15, 602–613 (2016).
Hamedani, S. Y. et al. Ras-like without CAAX 2 (RIT2): a susceptibility gene for autism spectrum disorder. Metab. Brain Dis. 32, 751–755 (2017).
Bouquillon, S. et al. A 5.3Mb deletion in chromosome 18q12.3 as the smallest region of overlap in two patients with expressive speech delay. Eur. J. Med. Genet. 54, 194–197 (2011).
Bossers, K. et al. Analysis of gene expression in Parkinson’s disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol. 19, 91–107 (2009).
Wang, Q. et al. Single-cell transcriptomic atlas of the human substantia nigra in Parkinson’s disease. bioRxiv, 2022.2003.2025.485846, https://doi.org/10.1101/2022.03.25.485846 (2022).
Obergasteiger, J. et al. The small GTPase Rit2 modulates LRRK2 kinase activity, is required for lysosomal function and protects against alpha-synuclein neuropathology. NPJ Parkinsons Dis. 9, 44 (2023).
Sweeney, C. G. et al. Conditional, inducible gene silencing in dopamine neurons reveals a sex-specific role for Rit2 GTPase in acute cocaine response and striatal function. Neuropsychopharmacology 45, 384–393 (2020).
Sato, H., Kato, T. & Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev. Neurosci. 24, 115–123 (2013).
Nichols, R. J. LRRK2 Phosphorylation. Adv. Neurobiol. 14, 51–70 (2017).
Casiraghi, A. et al. Methylphenidate Analogues as a New Class of Potential Disease-Modifying Agents for Parkinson’s Disease: Evidence from Cell Models and Alpha-Synuclein Transgenic Mice. Pharmaceutics 14, 1595 (2022).
Lou, J. S. Fatigue in Parkinson’s disease and potential interventions. NeuroRehabilitation 37, 25–34 (2015).
Eshleman, A. J. et al. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J. Pharmacol. Exp. Ther. 289, 877–885 (1999).
Fahn, S. The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov. Disord. 23, S497–508 (2008).
Augustin, S. M., Loewinger, G. C., O’Neal, T. J., Kravitz, A. V. & Lovinger, D. M. Dopamine D2 receptor signaling on iMSNs is required for initiation and vigor of learned actions. Neuropsychopharmacology 45, 2087–2097 (2020).
Lemos, J. C. et al. Enhanced GABA Transmission Drives Bradykinesia Following Loss of Dopamine D2 Receptor Signaling. Neuron 90, 824–838 (2016).
Roberts, H. C. et al. The Association of Grip Strength With Severity and Duration of Parkinson’s: A Cross-Sectional Study. Neurorehabil. Neural. Repair 29, 889–896 (2015).
Jeyasingham, R. A., Baird, A. L., Meldrum, A. & Dunnett, S. B. Differential effects of unilateral striatal and nigrostriatal lesions on grip strength, skilled paw reaching and drug-induced rotation in the rat. Brain Res. Bull. 55, 541–548 (2001).
Cheng, H. C., Ulane, C. M. & Burke, R. E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725 (2010).
Shi, G. X. & Andres, D. A. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol. Cell Biol. 25, 830–846 (2005).
Cramb, K. M. L., Beccano-Kelly, D., Cragg, S. J. & Wade-Martins, R. Impaired dopamine release in Parkinson’s disease. Brain 146, 3117–3132 (2023).
Decressac, M., Volakakis, N., Bjorklund, A. & Perlmann, T. NURR1 in Parkinson disease–from pathogenesis to therapeutic potential. Nat. Rev. Neurol. 9, 629–636 (2013).
Kadkhodaei, B. et al. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc. Natl. Acad. Sci. USA 110, 2360–2365 (2013).
Wu, S. et al. The Dopamine Transporter Recycles via a Retromer-Dependent Postendocytic Mechanism: Tracking Studies Using a Novel Fluorophore-Coupling Approach. J. Neurosci. 37, 9438–9452 (2017).
Choy, R. W. et al. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 82, 55–62 (2014).
Temkin, P. et al. The Retromer Supports AMPA Receptor Trafficking During LTP. Neuron 94, 74–82 e75 (2017).
Jia, C. et al. alpha-Synuclein Negatively Regulates Nurr1 Expression Through NF-kappaB-Related Mechanism. Front. Mol Neurosci 13, 64 (2020).
Nonnekes, J. et al. Unmasking levodopa resistance in Parkinson’s disease. Mov. Disord. 31, 1602–1609 (2016).
van Rumund, A. et al. Peripheral decarboxylase inhibitors paradoxically induce aromatic L-amino acid decarboxylase. NPJ Parkinsons Dis 7, 29 (2021).
Burger, C., Nguyen, F. N., Deng, J. & Mandel, R. J. Systemic mannitol-induced hyperosmolality amplifies rAAV2-mediated striatal transduction to a greater extent than local co-infusion. Mol. Ther. 11, 327–331 (2005).
Wertman, V., Gromova, A., La Spada, A. R. & Cortes, C. J. Low-Cost Gait Analysis for Behavioral Phenoty** of Mouse Models of Neuromuscular Disease. J. Vis. Exp. https://doi.org/10.3791/59878 (2019).
Wang, Q. et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 10, 5234 (2019).
Acknowledgements
These studies were supported by R01DA015169 (H.E.M.), R01DA035224 (H.E.M.), F31DA045446 (P.J.K), 5T32AG49688 (M.L.), P20NS123220 (Z.Y.), R21NS109895 (Z.Y.), and the Parkinson’s Foundation Research Center (PF-RC-936279; Z.Y).
Author information
Authors and Affiliations
Contributions
P.J.K, H.E.M., Y.Z., Y.T., S.A.S., and Z.Y, designed the studies; P.J.K, Y.Z., M.L., E.K, Y.T.,R.F., R.P, and T.C. acquired data; P.J.K, H.E.M., R.G.P., and R.R.F. analyzed data; P.J.K, Z.Y, Y.Z., Y.T., S.A.S., and H.E.M. wrote the manuscript. All authors read, edited and approved of the submitted manuscript, and were accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kearney, P.J., Zhang, Y., Liang, M. et al. Silencing Parkinson’s risk allele Rit2 sex-specifically compromises motor function and dopamine neuron viability. npj Parkinsons Dis. 10, 41 (2024). https://doi.org/10.1038/s41531-024-00648-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00648-8
- Springer Nature Limited