

Abstract
The [1,2]- and [2,3]-Stevens rearrangements are one of the most fascinating chemical bond reorganization strategies in organic chemistry, and they have been demonstrated in a wide range of applications, representing a fundamental reaction tactic for the synthesis of nitrogen compounds in chemical community. However, their applicabilities are limited by the scarcity of efficient, general, and straightforward methods for generating ammonium ylides. Herein, we report a general difluorocarbene-induced tertiary amine-involved [1,2]- and [2,3]-Stevens rearrangements stemmed from in situ generated difluoromethyl ammonium ylides, which allows for the rearrangements of versatile tertiary amines bearing either allyl, benzyl, or propargyl groups, resulting in the corresponding products in one reaction under the same reaction conditions with a general way. Broad substrate scope, simple operation, mild reaction conditions and late-stage modification of natural products highlight the advantages of this strategy, meanwhile, this general rearrangement reaction is believed to bring opportunities for the transformations of nitrogen ylides and the assembly of valuable tertiary amines and amino acids. This will further enrich the reaction repertoire of difluorocarbene species, facilitate the development of reactions involving difluoromethyl ammonium salts, and provide an avenue for the development of this type of rearrangement reactions.
Similar content being viewed by others
Introduction
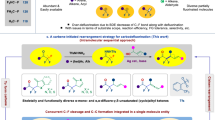
Tertiary amines and their derivatives are widely present in natural products and bioactive molecules as an important functionality, and it is beneficial to achieve the precise late-stage modification of existing drugs and natural products, without significantly altering the parental scaffolds and maintaining the functionalities1,2,3,4,5. The Stevens rearrangement is typically applied to construct complex nitrogen/sulfur-containing compounds through [1,2]- or [2,3]-sigmatropic rearrangement, starting from ammonium or sulfonium salts. However, harsh conditions, such as the presence of a strong base, are usually necessary and unavoidable, and the key intermediate is nitrogen6,7,8,9,10,11,12,13,14 or sulfur15,16,17,18,19 ylide. Therefore, Stevens rearrangement is often considered a commonly used molecular editing method for prevalent tertiary amines. Usually, the methods of producing nitrogen ylides mainly include the following strategies: prepared from tertiary amines within situ-generated arynes20,21,22 or alkyl halides, or activated by equivalent of Lewis acids23,24,25,26,27 (Fig. 1A). For [1,2]-Stevens rearrangement, it typically involves the migration of benzyl substituents on tertiary amines to the adjacent carbon atom through an [1,2]-sigmatropic rearrangement mechanism28,29, or the ring expansion reactions of small cyclic amines with diazo compounds, as reported by Lacour and Feng et al.30,31,32. For [2,3]-sigmatropic rearrangement, transition-metal-catalyzed methods were also developed for the generation of nitrogen ylides33,34,35,36,37,38,39,40, for instance, a Pd-catalyzed allylic amination strategy using tertiary amino esters and allyl carbonates for the generation of nitrogen ylides has been disclosed by Tambar group in 2011. These reactive intermediates (arynes20,21,22, π-Allyl palladium complex36, etc.) enable the Stevens rearrangement to proceed under milder conditions, avoiding the harsh conditions such as high temperature and strong base required by the reaction previously. Tertiary allyl amines with diazoesters via a Rh-catalyzed cross-coupling for direct access to nitrogen ylides have emerged as a universal method. In 2021, Zhang and coworkers demonstrated a 5-endo-dig cyclization of tertiary amines with intramolecular alkynes activated by π-Lewis-acid to form quaternary ammonium salts, then render nitrogen ylides41. However, the universalities of the known Stevens rearrangements are quite poor, usually it is difficult for them to be compatible with all allyl, propargyl, and benzyl groups in one reaction under the same conditions6,7,8,9,10,11,12,13,14, which thus restricts the derivation of product diversity. Consequently, studies toward the efficient and versatile methods for both [1,2]- and [2,3]-Stevens rearrangements are highly desirable.

A Previous strategies of constructing ammonium ylides for [1,2]- or [2,3]-Stevens rearrangement. B Our hypothesis: could difluorocarbene enable Stevens rearrangement of tertiary amines? C Difluorocarbene-induced both [1,2]- and [2,3]-Stevens rearrangement via difluoromethyl ammonium ylides (this work).
Given the narrow substrate scopes and limited migration types in the previous strategies, we envisioned discovering an efficient and powerful approach, which can start with simple starting materials to achieve both [1,2]-and [2,3]-Stevens rearrangements and be compatible with all of allyl, propargyl, and benzyl groups smoothly in the same reaction conditions. Based on the structure of ammonium ylides10,11,12,13,14,15, we focus our attention on the difluoromethyl ammonium salts, which is inspired by our previous research on the reactivity of difluorocarbene42,43. Since this structure was proposed by our group in 202044, it has been extensively studied, and this key intermediate can be used to achieve the construction of complex fluorinated compounds45,46,47,48, the synthesis of natural products49,50, and deaminative Suzuki cross-coupling reactions without transition-metal catalysis51. Based on its exceptional performance and enormous application potential, we envisioned whether [1,2]- and [2,3]-Stevens rearrangement reactions could be performed on the special in situ-generated difluoromethyl ammonium ylide I in the presence of base, since this ylide I could be readily converted to a difluoromethyl-bearing nitrogen ylide II with water52,53. Moreover, since the difluoromethyl quaternary ammonium ylide II54,55 also exhibits electron-withdrawing characteristics, this property may potentially facilitate Stevens rearrangement even more smoothly. However, there are some inherent challenges in this hypothesis: (a) When substituents with large steric hindrance are on tertiary amines, it might be difficult for nitrogen atom to capture difluorocarbene species, resulting in the failure of the reaction. (b) When difluoromethyl ammonium salt contains allyl or benzyl groups, an SN2 pathway might take place by the nucleophilic reagents (for instance, halogen ions) in the system, resulting in the formation of benzyl or allyl halides, thus delaying the occurrence of Stevens rearrangement. (c) Difluoromethyl ammonium salt might convert into a formylation product56,57 in the presence of water, thereby preventing the rearrangement reaction from occurring (Fig. 1B). To our delight, when we used difluorocarbene species to induce Stevens rearrangement, we could smoothly obtain the expected product, and it effectively avoids the occurrence of the aforementioned side reactions. Herein, we report an efficient difluorocarbene-induced Stevens rearrangement reaction. Mild reaction conditions, wide substrate range, diverse migration modes, multiple product types, and the extensive modification of various natural products all convincingly demonstrate the advantages of this method. This will further enrich the reaction repertoire of difluorocarbene species, facilitate the development of reactions involving difluoromethyl ammonium salts, and provide an avenue for the development of this type of rearrangement reaction. In such a difluorocarbene-induced rearrangement reaction, it can accommodate various types of rearrangements with different substrates, significantly expanding the utility and product diversity of this reaction. When the substituent on nitrogen atom is a benzyl group, [1,2]-rearrangement proceeds smoothly. When the substituent is an alkyne or an alkene, [2,3]-sigmatropic rearrangement can occur efficiently, and after alkyne rearrangement, 1,1-disubstituted allenes are readily obtained (Fig. 1C).
Results
Reaction optimization
To test the feasibility of the hypothesis, we set out to probe the reaction conditions using tertiary amine 1a, BrCF2COOK (2a) as tertiary amine activating reagent, K2CO3 as the base in the presence of CH3CN (Table 1). The desired Stevens rearrangement product 3a was obtained in 45% isolated yield. Encouraged by the above results, we then proceeded to further optimize the reaction conditions, types, and dosages of difluorinated reagents, bases, and reaction temperatures on the product yields, were emphatically investigated in Table 1. We examined originally the effects of different difluorocarbene reagents on this reaction, the results indicated that among BrCF2COOK (2a), BrCF2COONa (2b), BrCF2COOEt (2c), ClCF2COONa (2d), ClCF2COONa (2e), ClCF2COONa (2d) demonstrated the best performance and the desired product 3a was obtained in 51% yield (entries 1–5, Table 1). Subsequently, further investigations were conducted on the effects of different types of bases on the difluorocarbene-induced Stevens rearrangement, which suggested that K3PO4 exhibited the best overall balance of activity and basicity among HCOONa, Rb2CO3, LiOH, Na2CO3, Et3N, as well as K3PO4 (entries 6–11, Table 1), the yield of 3a increased to 85%. The dosage of difluorinated reagents and base were also assessed (entries 12–16, Table 1), a better outcome was obtained when 1.5 equiv of ClCF2COONa (2d) was employed instead of 3 equiv. Base evaluations suggested that 3 equiv. of K3PO4 is necessary to ensure the high yield of this rearrangement reaction (see Supplementary Information for details).
Substrate scope
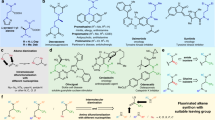
After identifying the optimal conditions, we turned to evaluate the substrate scope for this difluorocarbene-induced Stevens rearrangement reaction (Fig. 2). We investigated the effect of ester groups as electron-withdrawing groups on the rearrangement reaction. Different alcohol-derived esters including methyl, ethyl, tert-butyl and some other alkyl groups were all well tolerated in this rearrangement reaction, and afforded the corresponding coupling products 3a–3f in 68-85% yields, respectively. Different benzyl as the ester derivatives were also compatible substituents for this transformation (1g-1o), the corresponding product (3g-3o) was obtained in good to excellent yields. Notably, this protocol also featured an admirable scope with respect to ketone substrates. The corresponding target products 3p-3z were all smoothly delivered (69–86% yields), no matter that it was an aliphatic ketone or an aryl ketone. Aliphatic ketones, such as cyclopropyl (1p), tert-butyl (1q) and adamantly (1r) were all delivered the corresponding rearrangement products (3p-3r) with a moderate to excellent yields. For the aromatic ones, substrates bearing electron-neutral (1s-1t), electron-rich (1u), as well as electron-deficient substituents (1v) at the para-position of the aromatic rings all furnished the desired rearrangement products (3s-3v) smoothly. Of note, this reaction was even not greatly disturbed when a free hydroxyl group at the para-position of the aromatic rings, the final product 3w still could be obtained in decent yield. In addition, the meta-methoxy substrate 1x is also a good substrate for this transformation. This study was also easily extendable to the heteroaromatic ketone such as thiophene (3y). Fused ring reactant like 1-naphthaldehyde (1z) was also a suitable candidate for this transformation and the corresponding product 3z was obtained in 69% yield. We also made attempts with other electron-withdrawing groups and found that both cyano and phosphate esters were compatible with our reaction system. They afforded the allylic rearranged products with yields of 56% and 31%, respectively. When employing an amide as the electron-withdrawing group, it failed to induce the Stevens rearrangement reaction. Instead, we observed the formation of a product where the tertiary amine undergoes direct allylic cleavage at the nitrogen atom upon interaction with difluorocarbene, followed by formylation.

Reaction condition: a1 (1 equiv., 0.2 mmol), 2d (1.5 equiv., 0.3 mmol), H2O (5 equiv.), K3PO4 (3 equiv.), CH3CN (2 mL) at 90 °C for 12 h under argon. ND not detected.
Encouraged by the above results, we further explored the tolerance range of amine substrates. Since many amino acid derivatives typically include a polysubstituted skeleton, we then wondered whether this Stevens rearrangement can also be employed for such poly-substituted amino esters. Not surprisingly, when these substrates were subjected to the previous standard conditions it was found that the corresponding target products could only be obtained in 25% yield. We replaced Rb2CO3 as the base and found that the amount of water had a significant impact on the reaction outcomes (See Supplementary Table 5 in Supplementary Information for details). After simple condition reevaluations, expectedly, these substrates were equipotential to afford the corresponding modified amino ester derivatives in moderate to excellent yields under the marginally reoptimized conditions (Fig. 3). For such polysubstituted amino esters, different esters, including both aliphatic and benzylic ones all smoothly achieved this transformation in decent yields (6a-6d). For cyclic amino esters, like lactone (4e) and derivatives of proline (4f-4h), it suggests that they were all suitable candidates for this transformation and the corresponding polysubstituted amino ester derivatives (6e-6h) were obtained smoothly. More importantly, when internal alkene 4i was used as a substrate, it underwent the Stevens rearrangement to yield the terminal alkene product 6i, which provides further evidence on the occurrence of a [2,3]-sigmatropic rearrangement process in this reaction. Of note and fortunately, benzyl group was also compatible with the difluorocarbene-induced rearrangement reaction, resulting in benzyl [1,2]-sigmatropic rearrangement of ammonium ylides. Next, N-benzyl with various substituents on the benzene ring attached to the amino esters was explored (5a-5e), various functional groups were tolerable in the reaction, and delivered the corresponding products 7a-7e in decent yields.

Reaction condition: a4/5 (1 equiv., 0.2 mmol), 2d (1.5 equiv., 0.3 mmol), H2O (15 equiv.), Rb2CO3 (3 equiv.), CH3CN (2 mL) at 90 °C for 12 h under argon.
After examining the substrate scope of allyl and benzyl Stevens rearrangement reactions, we then set out to evaluate the generality of this protocol for propargyl tertiary amines (Fig. 4). It was found that esters bearing variations on the N-propargyl units were perfectly compatible with the reaction conditions, which further indicates that the reaction has a wide range of substrate tolerance. Notably, an N-propargyl substrate with a terminal acidic proton (i.e., R = H) also underwent a smooth conversion to the desired product 9a in 82% yield. And for the tert-butyl substituted amino ester 8b, it could also be able to deliver the corresponding products 9b in a good yield. Meanwhile, the propargyl substrates, which bear different alkyl and aryl groups were also good candidates for the rearrangement. For instance, this reaction worked well with methyl, ethyl, cycloalkyl, amyl, and tert-butyl-substituted substrates (8c-8g), giving the corresponding products 9c-9g in moderate to good yields. Alkyne derivatives substituted with pyran (8 h) and piperidine (8i) were also suitable substrates, which can also render the corresponding products 9h–9i in the yields of 77% and 62%, respectively. For alkyne compounds containing unprotected alkyl alcohols (8j), the final product 9j was successfully obtained without yield erosion, which also indicates that the reaction has good functional group tolerance. For 1, 3-enyne compounds, they were also good substrates for this rearrangement reaction, both terminal alkene 8k and internal cyclic alkene 8l could be smoothly converted into the target products 9k-9l with good yields accordingly. N-Propargyl substrates bearing electron-neutral (-H), electron-donating (-Et, and -OMe), and electron-withdrawing (-Cl, -Br, and -CHO) groups at the para position of benzene rings all furnished the desired allene products (9m-9r) smoothly. Of note, the structure of compound 9p was unambiguously confirmed by X-ray crystal analysis.

Reaction condition: a8 (1 equiv., 0.2 mmol), 2d (3 equiv., 0.6 mmol), H2O (15 equiv.), Rb2CO3 (3 equiv.), CH3CN (2 mL) at 90 oC for 12 h under argon.
Tertiary amines are also the core backbone of many natural product molecules. The above results clearly demonstrated that our method has a very broad substrate scope and wide functional group compatibility. Subsequently, we then focused on exploring the substrate tolerance range for the late-stage modification of natural products and bioactive molecules in this reaction (Fig. 5). Encouragingly, a series of natural products (Leaf alcohol, β-Rhodinol, Geraniol, Fenchol, L(-)-Borneol, DL-Menthol, Diacetone-D-glucose) were all derivatized into the corresponding amino esters and installed into our substrates (10a–10g) which, upon treatment with ClCF2COONa (2d) under the optimized standard conditions, were all smoothly incorporated into the eventual Stevens rearrangement products (11a–11g), which showcased the viability of employing this protocol for a late-stage functionalization of complex amines.

Reaction condition: a10 (1 equiv., 0.2 mmol), 2d (1.5 equiv., 0.3 mmol), H2O (5 equiv.), K3PO4 (3 equiv.), CH3CN (2 mL) at 90 °C for 12 h under argon.
Synthetic utility
To demonstrate the synthetic utility of this difluorocarbene-induced Stevens rearrangement protocol, various post-functionalizations were employed (Fig. 6). The scalability was demonstrated by the successful gram-scale reaction of 3p, 7d and 9o, and the corresponding products were generated without a significant decrease in yields, which further elucidates the practical value of the Stevens rearrangement reaction (Fig. 6a). Furthermore, the olefin products are highly versatile synthetic intermediates that can be readily transformed to an alkane in the presence of Pd/C and H258, moreover, under the influence of Pd/C and H2, the three-membered ring was simultaneously opened to yield a straight-chain propane 12. The formamide product of this difluorocarbene-induced Stevens rearrangement can also be hydrolyzed with concentrated sulfuric acid59 to obtain a secondary amine 13. These scale-up experiments and the corresponding transformations of rearrangement products were successfully achieved, demonstrating the utility of the reaction and the diverse and useful transformations of the products.

a Reactions performed at gram-scale. b Derivatizations of Stevens rearrangement products. Reaction conditions: a1p (1 equiv., 5 mmol), 2d (1.5 equiv., 7.5 mmol), H2O (5 equiv.), K3PO4 (3 equiv.), CH3CN (15 mL) at 90 °C for 18 h under argon. b5d or 8o (1 equiv., 5 mmol), 2d (1.5 equiv., 7.5 mmol), H2O (15 equiv.), Rb2CO3 (3 equiv.), CH3CN (2 mL) at 90 °C for 18 h under argon.
Mechanistic considerations
Based on previous literature60,61 and experimental outcomes from this work, we speculate the possible mechanism of this difluorocarbene-induced tertiary amine-involved [1,2]- and [2,3]-Stevens rearrangement reactions (Fig. 7). The reaction first generates difluorocarbene in situ through the decomposition of difluoroalkylative reagent 2d, which then combines with tertiary amine A to form the key active quaternary ammonium salt intermediate B. Direct completion of the [1,3]-proton shift via a strained four-membered ring transition state from B to C is very challenging based on the work proposed by Yu and colleagues in 201752,53, therefore, we think that protonation and deprotonation assisted by water are more feasible. The difluoromethyl carbanion on the quaternary ammonium salt B might be hydrolyzed in situ to lead to an amidyl species during this process under the action of base and water. Subsequent [2,3]- or [1,2]-sigmatropic rearrangement of C gives the corresponding rearrangement products through either concerted mechanism (for [2,3]-sigmatropic rearrangement) or radical pair path (for [1,2]-sigmatropic rearrangement) respectively6,7,8,9,10,11,12,13,14.

Possible reaction mechanism of difluorocarbene-induced [1,2]- and [2,3]-Stevens rearrangement of tertiary amines.
In summary, we have reported a difluorocarbene-induced Stevens rearrangement reaction. Within this reaction system, various types of Stevens rearrangements could be accommodated, including the [2,3]-sigmatropic rearrangement of allylic and propargyl tertiary amines, as well as the [1,2]-sigmatropic rearrangement of benzylic tertiary amines. Additionally, a variety of migration products can be obtained efficiently. More importantly, the smooth conversion of this reaction further broadens the application of difluoromethyl ammonium salts, enabling Stevens rearrangement reactions beyond C-N bond activation, nucleophilic reagents, and deaminative cross-coupling. Given the simplicity and efficiency of these reactions, it is anticipated that this strategy would open up avenues to facilitate the exploration of such rearrangement reactions and contribute to the study of the properties of difluoromethyl ammonium salts.
Methods
Synthesis of 3
In air, tertiary amines 1 (1 eq, 0.2 mmol), ClCF2COONa (1.5 eq, 0.3 mmol), and K3PO4 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a magnetic stirring bar. The vessel was evacuated and filled with argon (three cycles). CH3CN (2 mL) and H2O (5 eq, 1 mmol) were added by syringe under argon atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 12 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography (silica gel, petroleum ether: EtOAc = 2:1, v/v) to give the desired products.
Synthesis of 6/7
In air, allylic amines 4 or benzyl amines 5 (1 eq, 0.2 mmol), ClCF2COONa (1.5 eq, 0.3 mmol), and Rb2CO3 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a magnetic stirring bar. The vessel was evacuated and filled with argon (three cycles). CH3CN (2 mL) and H2O (15 eq, 3 mmol) were added by syringe under argon atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 12 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography (silica gel, petroleum ether: EtOAc = 2:1, v/v) to give the desired products.
Synthesis of 9
In air, propargyl tertiary amines 8 (1 eq, 0.2 mmol), ClCF2COONa (1.5 eq, 0.3 mmol), and Rb2CO3 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a magnetic stirring bar. The vessel was evacuated and filled with argon (three cycles). CH3CN (2 mL) and H2O (15 eq, 3 mmol) were added by syringe under argon atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 12 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography (silica gel, petroleum ether: EtOAc = 2:1, v/v) to give the desired products.
Data availability
Data relating to the materials and methods, optimization studies, experimental procedures, NMR spectra, and mass spectrometry are available in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2305294 (9p). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Data can also be obtained from the corresponding author upon request.
References
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Coleman et al. Investigation of the binding determinants of phosphopeptides targeted to the src homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 48, 6661–6670 (2005).
Corma, A., Iborra, S. & Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107, 2411–2502 (2007).
Meusel, M. & Gütschow, M. Recent developments in hydantoin chemistry. A review. Org. Prep. Proced. Int. 36, 391–443 (2004).
Ouyang, K., Hao, W., Zhang, W.-X. & **, Z. Transition-metal-catalyzed cleavage of C-N single bonds. Chem. Rev. 115, 12045–12090 (2015).
Wicker, G., Zhou, R., Schoch, R. & Paradies, J. Sigmatropic [1,5] carbon shift of transient c3 ammonium enolates. Angew. Chem. Int. Ed. 61, e202204378 (2022).
Vanecko, J. A., Wan, H. & West, F. G. Recent advances in the stevens rearrangement of ammonium ylides. Application to the synthesis of alkaloid natural products. Tetrahedron 62, 1043–1062 (2006).
Thomson, T. & Stevens, T. S. 263. Degradation of quaternary ammonium salts. Part VII. New cases of radical migration. J. Chem. Soc. 1932–1940 (1932).
Tayama, E. Recent advances in the base-induced sommelet-hauser rearrangement of amino acid derived ammonium ylides. Chem. Rec. 15, 789–800 (2015).
Stevens, T. S., Snedden, W. W., Stiller, E. T. & Thomson, T. Degradation of quaternary ammonium salts. Part III. J. Chem. Soc. 2119–2125 (1930).
Stevens, T. S. Degradation of quaternary ammonium salts. Part II. J. Chem. Soc. 2107–2119 (1930).
Shi, C.-Y., Zhou, B., Teng, M.-Y. & Ye, L.-W. Recent advances in asymmetric [1,2]-stevens-type rearrangement via metal carbenes. Synthesis 55, 2118–2127 (2023).
Sheng, Z., Zhang, Z., Chu, C., Zhang, Y. & Wang, J. Metal-catalyzed [2,3]-sigmatropic rearrangements of ylides: an update of the most recent advances. Tetrahedron 73, 4011–4022 (2017).
Schwartz, Z., Valiton, C., Lovasz, M. & Roberts, A. G. Recent applications of ammonium ylide based [2,3]-sigmatropic and [1,2]-stevens rearrangements to transform amines into natural products. Synthesis 56, 87–106 (2023).
Zhang, Z. et al. Catalytic asymmetric trifluoromethylthiolation via enantioselective [2,3]-sigmatropic rearrangement of sulfonium ylides. Nat. Chem. 9, 970–976 (2017).
Shi, C.-Y., Zhou, B., Teng, M.-Y. & Ye, L.-W. Recent advances in the asymmetric doyle-kirmse reaction. Synthesis 55, 3895–3905 (2023).
Li, L., Chen, B., Chen, J. & Huang, Y. Enantioselective intramolecular [2,3]-sigmatropic rearrangement of aldehydes via a sulfonium enamine intermediate. Angew. Chem. Int. Ed. 59, 20904–20908 (2020).
Hu, L., Li, J., Zhang, Y., Feng, X. & Liu, X. Enantioselective [1,2]-Stevens rearrangement of thiosulfonates to construct dithio-substituted quaternary carbon centers. Chem. Sci. 13, 4103–4108 (2022).
Thangraraj, M., Gayker, R., Roy, T. & Biju, A. Synthesis of functionalized β-keto arylthioethers by the aryne induced [2,3] stevens rearrangement of allylthioethers. J. Org. Chem. 82, 4470–4476 (2017).
Zhang, J. et al. Aryne-mediated [2,3]-sigmatropic rearrangement of tertiary allylic amines. Org. Lett. 18, 4872–4875 (2016).
Roy, T. et al. The aryne [2,3] stevens rearrangement. Org. Lett. 18, 5428–5431 (2016).
Zhou, M.-G., Dai, R.-H. & Tian, S.-K. Nucleophilic addition of tertiary propargylic amines to arynes followed by a [2,3]-sigmatropic rearrangement. Chem. Commun. 54, 6036–6039 (2018).
Workman, J. A. et al. Asymmetric [2,3]-rearrangement of glycine-derived allyl ammonium ylids. J. Am. Chem. Soc. 127, 1066–1067 (2005).
West, T. H., Daniels, D. S. B., Slawin, A. M. Z. & Smith, A. D. An isothiourea-catalyzed asymmetric [2,3]-rearrangement of allylic ammonium ylides. J. Am. Chem. Soc. 136, 4476–4479 (2014).
Chen, Z.-Y. et al. Perfluorobutyl iodide mediated [1,2] and [2,3] stevens rearrangement for the synthesis of indolin-3-ones. Adv. Synth. Catal. 362, 4368–4372 (2020).
Dai, R.-H., Han, L., Wang, Q. & Tian, S.-K. Strain-release C-C bond cleavage enables the [2,3]-sigmatropic rearrangement of tertiary allylamines. Chem. Commun. 54, 8449–8451 (2021).
**, Y.-X., Yu, B.-K., Qin, S.-P. & Tian, S.-K. Epoxide-mediated stevens rearrangements of α-amino-acid-derived tertiary allylic, propargylic, and benzylic amines: convenient access to polysubstituted morpholin-2-ones. Chem. Eur. J. 25, 5169–5172 (2019).
Tuzina, P. & Somfai, P. Asymmetric lewis acid mediated [1,2]-rearrangement of proline-derived ammonium ylides. Org. Lett. 11, 919–921 (2009).
Glaeske, K. W. & West, F. G. Chirality transfer from carbon to nitrogen to carbon via cyclic ammonium ylides. Org. Lett. 1, 31–34 (1999).
Wang, K. et al. Asymmetric catalytic ring-expansion of 3-methyleneazetidines with α-diazo pyrazoamides towards proline-derivatives. Angew. Chem. Int. Ed. 62, e202307249 (2023).
Miller, D. C., Lal, R. G., Marchetti, L. A. & Arnold, F. H. Biocatalytic one-carbon ring expansion of aziridines to azetidines via a highly enantioselective [1,2]-stevens rearrangement. J. Am. Chem. Soc. 144, 4739–4745 (2022).
Hong, F.-L. et al. Copper-catalyzed asymmetric diyne cyclization via [1,2]-stevens-type rearrangement for the synthesis of chiral chromeno[3,4-c]pyrroles. Angew. Chem. Int. Ed. 61, e202115554 (2022).
Yu, X., Wannenmacher, N. & Peters, R. Stereospecific asymmetric synthesis of tertiary allylic alcohol derivatives by catalytic [2,3]-meisenheimer rearrangements. Angew. Chem. Int. Ed. 59, 10944–10948 (2020).
**, S. et al. Generation and [2,3]-sigmatropic rearrangement of ammonium ylides from cyclopropyl ketones for chiral indolizidines with bridgehead quaternary stereocenters. Org. Lett. 24, 6957–6961 (2022).
Spoehrle, S. S. M., West, T. H., Taylor, J. E., Slawin, A. M. Z. & Smith, A. D. Tandem palladium and isothiourea relay catalysis: enantioselective synthesis of α-amino acid derivatives via allylic amination and [2,3]-sigmatropic rearrangement. J. Am. Chem. Soc. 139, 11895–11902 (2017).
Soheili, A. & Tambar, U. K. Tandem catalytic allylic amination and [2,3]-stevens rearrangement of tertiary amines. J. Am. Chem. Soc. 133, 12956–12959 (2011).
Schmid, S. C., Guzei, I. A., Fernández, I. & Schomaker, J. M. Ring expansion of bicyclic methyleneaziridines via concerted, near-barrierless [2,3]-stevens rearrangements of aziridinium ylides. ACS Catal. 8, 7907–7914 (2018).
Nickerson, L. A. et al. Enantioselective synthesis of isochromans and tetrahydroisoquinolines by C-H insertion of donor/donor carbenes. Chem. Sci. 11, 494–498 (2020).
Lin, Q. et al. Chiral N,N′-dioxide/Mg(OTf)2 complex-catalyzed asymmetric [2,3]-rearrangement of in situ generated ammonium salts. Chem. Sci. 11, 3068–3073 (2020).
Bao, H., Qi, X. & Tambar, U. K. Catalytic enantioselective [2,3]-rearrangements of amine N-oxides. J. Am. Chem. Soc. 133, 1206–1208 (2011).
**, S. et al. Lewis acid-catalyzed domino generation/[2,3]-sigmatropic rearrangement of ammonium ylides to access chiral azabicycles. Sci. Adv. 7, eabd5290 (2021).
Ma, X., Su, J. & Song, Q. Unconventional transformations of difluorocarbene with amines and ethers. Acc. Chem. Res. 56, 592–607 (2023).
Ma, X. & Song, Q. Recent progress on selective deconstructive modes of halodifluoromethyl and trifluoromethyl-containing reagents. Chem. Soc. Rev. 49, 9197–9219 (2020).
Su, J., Ma, X., Ou, Z. & Song, Q. Deconstructive functionalizations of unstrained carbon-nitrogen cleavage enabled by difluorocarbene. ACS Cent. Sci. 6, 1819–1826 (2020).
Zhang, G. et al. Atom recombination of difluorocarbene enables 3-fluorinated oxindoles from 2-aminoarylketones. CCS Chem. 4, 1671–1679 (2021).
Su, J., Hu, X., Huang, H., Guo, Y. & Song, Q. Difluorocarbene enables to access 2-fluoroindoles from ortho-vinylanilines. Nat. Commun. 12, 4986 (2021).
Liu, A., Ni, C., **e, Q. & Hu, J. Transition-metal-free controllable single and double difluoromethylene formal insertions into c-h bonds of aldehydes with TMSCF2Br. Angew. Chem. Int. Ed. 62, e202217088 (2023).
Liu, A., Ni, C., **e, Q. & Hu, J. TMSCF2Br-enabled fluorination-aminocarbonylation of aldehydes: modular access to α-fluoroamides. Angew. Chem. Int. Ed. 61, e202115467 (2022).
Lim, H., Seong, S., Kim, Y., Seo, S. & Han, S. Biopatterned reorganization of alkaloids enabled by ring-opening functionalization of tertiary amines. J. Am. Chem. Soc. 143, 19966–19974 (2021).
Kim, Y., Heo, J., Kim, D., Chang, S. & Seo, S. Ring-opening functionalizations of unstrained cyclic amines enabled by difluorocarbene transfer. Nat. Commun. 11, 4761 (2020).
Su, J., Li, C., Hu, X., Guo, Y. & Song, Q. Deaminative arylation and alkenyaltion of aliphatic tertiary amines with aryl and alkenylboronic acids via nitrogen ylides. Angew. Chem. Int. Ed. 61, e202212740 (2022).
Wang, Y. & Yu, Z.-X. Sigmatropic proton shifts: a quantum chemical study. Org. Biomol. Chem. 15, 7439–7446 (2017).
Wang, Y., Cai, P.-J. & Yu, Z.-X. Carbanion translocations via intramolecular proton transfers: a quantum chemical study. J. Org. Chem. 82, 4604–4612 (2017).
Nawrot, E. & Joñczyk, A. Difluoromethyltrialkylammonium salts their expeditious synthesis from chlorodifluoromethane and tertiary amines in the presence of concentrated aqueous sodium hydroxide. The catalytic process. J. Org. Chem. 72, 10258–10260 (2007).
Mita, T., Harabuchi, Y. & Maeda, S. Discovery of a synthesis method for a difluoroglycine derivative based on a path generated by quantum chemical calculations. Chem. Sci. 11, 7569–7577 (2020).
Ma, X., Su, J., Zhang, X. & Song, Q. Chlorodifluoromethane as a C1 synthon in the assembly of N-containing compounds. iScience 19, 1–13 (2019).
Ma, X., Deng, S. & Song, Q. Halodifluoroacetates as formylation reagents for various amines via unprecedented quadruple cleavage. Org. Chem. Front. 5, 3505–3509 (2018).
Zhang, Z.-H. et al. Copper-catalyzed enantioselective Sonogashira-type oxidative cross-coupling of unactivated C(sp3)-H bonds with alkynes. Nat. Commun. 10, 5689 (2019).
Li, H., Qian, H.-F. & Feng, G. Diversity-oriented synthesis of azo disperse dyes with improved fastness properties via employing Ugi four-component reaction. Dyes Pigm. 165, 415–420 (2019).
Biswas, B. & Singleton, D. A. Controlling selectivity by controlling the path of trajectories. J. Am. Chem. Soc. 137, 14244–14247 (2015).
Biswas, B., Collins, S. C. & Singleton, D. A. Dynamics and a unified understanding of competitive [2,3]- and [1,2]-sigmatropic rearrangements based on a study of ammonium ylides. J. Am. Chem. Soc. 136, 3740–3743 (2014).
Acknowledgements
Financial support from the National Key R&D Program of China (2023YFF0723900), National Natural Science Foundation of China (21931013 and 22271105), Natural Science Foundation of Fujian Province (2022J02009), and Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Q.S. conceived and directed the project. J.S. and Y.G. performed experiments and prepared the supplementary information. C.L. helped collecting some new compounds and analyzing the data. Q.S. and J.S. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Akkattu Biju and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, J., Guo, Y., Li, C. et al. Difluorocarbene-induced [1,2]- and [2,3]-Stevens rearrangement of tertiary amines. Nat Commun 15, 4794 (2024). https://doi.org/10.1038/s41467-024-49054-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49054-x
- Springer Nature Limited