

Abstract
Currently, no oral medications are available for type 1 diabetes (T1D). While our recent randomized placebo-controlled T1D trial revealed that oral verapamil had short-term beneficial effects, their duration and underlying mechanisms remained elusive. Now, our global T1D serum proteomics analysis identified chromogranin A (CHGA), a T1D-autoantigen, as the top protein altered by verapamil and as a potential therapeutic marker and revealed that verapamil normalizes serum CHGA levels and reverses T1D-induced elevations in circulating proinflammatory T-follicular-helper cell markers. RNA-sequencing further confirmed that verapamil regulates the thioredoxin system and promotes an anti-oxidative, anti-apoptotic and immunomodulatory gene expression profile in human islets. Moreover, continuous use of oral verapamil delayed T1D progression, promoted endogenous beta-cell function and lowered insulin requirements and serum CHGA levels for at least 2 years and these benefits were lost upon discontinuation. Thus, the current studies provide crucial mechanistic and clinical insight into the beneficial effects of verapamil in T1D.
Similar content being viewed by others

Introduction
Diabetes continues to grow as a chronic global health problem, affecting people of all ages. Since the discovery of insulin, a century ago, therapies have improved dramatically, but many critical needs and hurdles remain and prevent subjects with diabetes from living a truly normal life. In the case of type 1 diabetes (T1D), which involves autoimmune and inflammatory processes and destruction of insulin-producing pancreatic beta cells, exogenous insulin is still the only available therapy, and it is associated with the inherent risk of low blood glucose levels or hypoglycemic episodes that can be life-threatening. In addition, administration of insulin entails either continuous infusion via a pump or multiple daily injections and no oral medications are available. Recently, we reported the results of a small, phase 2, randomized placebo-controlled trial, using oral verapamil, an approved blood pressure medication, in new-onset T1D subjects1. Subjects receiving verapamil had improved endogenous beta cell function (as measured by a 2 h mixed-meal-stimulated C-peptide area under the curve (AUC)), lower insulin requirements, and fewer hypoglycemic events as compared to individuals getting placebo added to their standard insulin regimen1. Even though all subjects were normotensive, verapamil did not lead to any hypotension or any adverse events. While highly promising, these findings also raised a number of new mechanistic questions, including what exact biological changes verapamil elicits in humans with T1D, how long they may last, and how these changes and any potential associated therapeutic success could be monitored. The current studies were aimed at addressing these questions using proteomics, transcriptomics, and pathophysiological approaches. In this work, we now show that verapamil reverses T1D associated increases in serum CHGA levels, proinflammatory interleukin-21 (IL-21) levels, and T-follicular-helper (Tfh) cell markers and promotes an anti-oxidative, anti-apoptotic and immunomodulatory gene expression profile in human islets. In addition, our results suggest that continuous use of oral verapamil in subjects with T1D may delay loss of beta cell function and lower insulin requirements for at least 2 years post-diagnosis and that such therapeutic success or disease progression can be monitored by changes in serum CHGA.
Results
To assess potential systemic changes in response to verapamil treatment, we conducted a global proteomics analysis using liquid chromatography-tandem mass spectrometry (LC-MS/MS) of serum samples from subjects at baseline and after 1 year of receiving verapamil or placebo and determined the effect of treatment on changes over time. 10 subjects had sufficient usable serum for LC-MS/MS at both of these time points resulting in 20 samples used for proteomics analysis (Supplementary Fig. 1). The baseline subject characteristics of this subset and of the full set of study participants are shown in Supplementary Table 1 and they demonstrate that there were no significant differences between the treatment groups with respect to age, gender, race, BMI, or HbA1C. Also, analysis of serum from the same subject before and 1 year after treatment allowed each subject to provide its own baseline control and avoid confounding effects from inter-individual variability. Applying a global serum proteomics workflow (Supplementary Fig. 1) along with this tightly controlled design, we were able to quantify 31,457 unique peptides and 867 proteins (<1% false discovery rate, FDR) with TMT reporter ion intensity data across all channels without missing data in the 20 serum samples (Supplementary Fig. 2). The raw dataset from this study is available at ProteomeXchange (accession # PXD026601). Following statistical analysis on treatment effect using a linear regression model, 53 proteins were identified whose relative abundance over time was significantly altered (P < 0.05) by verapamil treatment (Supplementary Table 2). Enrichr31,32.

RNA sequencing was performed on isolated human islets from three different individuals (A–C) treated for 24 h with or without verapamil (100 µM) with each donor serving as its own control. Volcano plot contains all genes with a baseMean expression of >500. Those genes with an adjusted DESeq2 P-value < 0.05 (calculated using a Wald test and the Benjamini–Hochberg method) are shown in blue (downregulated) and red (upregulated) (a). Key pathways modulated by differentially expressed genes (b). Heatmap showing key genes changed after treatment with verapamil (color scale represents log2 fold change) (c). Display of the normalized read counts for key genes before and after verapamil treatment of islets from each of the individual islet donors A–C (d).
Discussion
In summary, the results of these exploratory studies suggest that continuous use of oral verapamil in individuals with T1D may delay disease progression and lower insulin requirements for at least 2 years post-diagnosis and that this is associated with normalization of serum CHGA levels as well as of proinflammatory IL-21 levels and Tfh cell markers. In addition, they show that verapamil regulates the thioredoxin system and promotes an anti-oxidative, anti-apoptotic and immunomodulatory gene expression profile in human islets suggesting that together these protective changes might explain the overall beneficial effects observed with verapamil.
The significantly lower insulin requirements found after 2 years of verapamil treatment as compared to controls is consistent with earlier findings after 1 year of treatment1 and with the observed preservation of beta cell function. In addition, our results showing verapamil regulating the thioredoxin system and inhibiting TXNIP expression in the islets provide a potential mechanistic explanation for these beta cell sparing effects. This especially when considering the beta cell protective and anti-diabetic effects observed in different mouse models in response to genetic TXNIP deletion or verapamil-induced TXNIP inhibition26,29. Moreover, in humans with T1D even a small amount of preserved endogenous insulin production as opposed to higher exogenous insulin requirements has been shown to be associated with improved outcome33 and could help improve quality of life and lower the high costs associated with insulin use. The fact that these beneficial verapamil effects seemed to persist for 2 years, whereas discontinuation of verapamil led to disease progression, provides some additional support for this approach and its potential usefulness for long-term treatment. Intriguingly, global proteomic profiling of this unique set of before and after treatment samples also led to the discovery of serum CHGA as a potential therapeutic marker. Serum CHGA is a simple and easy blood test, showed good correlation with loss of beta cell function, accurately reflected changes in response to verapamil therapy or treatment discontinuation and as a major advantage, persisted over a time period of at least 2 years. Furthermore, due to its role as a T1D antigen8,9, it is tempting to speculate that the lowering of CHGA in response to verapamil might even help dampen some of the T1D autoimmunity. In any case, our results reveal for the first time that verapamil may reverse specific T1D-induced T-cell changes and thereby may also modulate certain aspects of the immune response. Intriguingly, Tfh cells and IL-21, both suggested to be downregulated by verapamil, have recently been reported to play an important role in the autoimmunity of T1D12,13,14. In fact, this may help explain why verapamil treatment was so successful even in the absence of any additional bona fide immunomodulatory intervention. This provocative idea of additional immune modulatory effects is supported by the observed enrichment in gene ontology processes associated with MHC class I antigen presentation and neutrophil-mediated functions in response to verapamil. Thus, the present exploratory studies provide the first indication of some potentially sustained benefits of verapamil use in the context of T1D. They further suggest that verapamil may result in protective effects not only at the level of insulin-producing islet beta cells, but also at the level of T-cells and proinflammatory cytokines, uncovering a previously unappreciated connection between verapamil use and the immune system in T1D. However, since the current studies were based on a very small subset of subjects, these initial findings will have to be confirmed in larger studies such as the ongoing Ver-A-T1D (NCT04545151) or CLVer (NCT04233034) verapamil trials. In addition, long-term trials with extensive sample analysis using this new knowledge will ultimately have to validate CHGA as a proper biomarker as well as the novel clinical and mechanistic insights gained in the current studies.
Methods
Human subjects
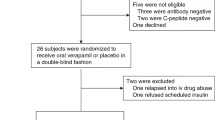
All human studies were approved by the University of Alabama at Birmingham (UAB) Internal Review Board and written informed consent was obtained from all participants in accordance with the criteria set by the Declaration of Helsinki. Participants’ compensation was $80 per completed MMTT. Subjects with T1D had been diagnosed within 3 months and were positive for T1D associated auto-antibodies. All continued on their standard insulin regimen but were on no other diabetes medications during the entire 2 years. They were taking randomly assigned verapamil (360 mg sustained-release daily) or placebo in a blinded fashion for one year as described in the protocol and consort table of the initial trial [clinicaltrials.gov/ct2/show/NCT02372253 2/20/2015]1 and then chose to be on or off verapamil and to be followed for a second year. This resulted in five subjects who continued to receive verapamil (360 mg sustained-release daily) for 2 years, four subjects who discontinued active study drug for the second year, and six participants randomized to the control arm who never received active study drug over the 2-year period. All other subjects declined the second-year follow-up and/or had no usable blood samples. The study subject demographic characteristics at baseline and at the start of year 2 are listed in Supplementary Tables 1 and 4, respectively. Supplementary Table 6 provides an additional overview of all the individual study participants, use of their samples, and their clinical data. Healthy volunteers were non-diabetic as confirmed by HbA1c, had not been diagnosed with any illness, were not receiving any prescription medications, and never received any study drug. Their characteristics are also shown in Supplementary Table 1 even though they only provided blood samples used as a comparison for some of the measurements of this exploratory study.
Remaining beta cell function was assessed using stimulated C-peptide AUC during a mixed-meal tolerance test (MMTT) as described previously4,5. The MMTT was only performed when fasting blood glucose levels were within the range of 3.9–11.1 mmol/L and otherwise, the test was rescheduled. Blood samples were collected at −10, 0, 15, 30, 60, 90, and 120 min for serum C-peptide. The mean C-peptide AUC (0–120 min) was calculated using the trapezoidal rule5 and the percent change from baseline was determined for each individual. In addition, the daily insulin dose required was calculated by analyzing the patient’s mean daily insulin use during a 2-week period at baseline, year 1 and year 2. Glycemic control was monitored by measurements of HbA1c. Serum samples and PBMCs were collected and stored at −80 °C until further analysis.
Proteomics/liquid chromatography-tandem mass spectrometry (LC-MS/MS)
20 serum samples from 10 subjects collected for each subject at baseline and after 1 year of receiving verapamil or placebo were analyzed using a standardized workflow (Supplementary Fig. 1) similarly as previously reported34. For sample processing, 40 µL of serum was subjected to immunodepletion of the most abundant serum proteins using a MARS Hu-14 column (Agilent Technologies, Palo Alto, CA) as previously described35. The flow-through fractions were concentrated using Amicon spin filters with 3 kDa molecular mass cutoffs (Millipore, Burlington, MA). Protein concentration was measured by BCA assay (Thermo Scientific, San Jose, CA) prior to protein digestion. The depleted samples were then digested by a urea-based protocol and the peptides were then desalted by solid-phase extraction (SPE) (Phenomenex, Torrance, CA) and dried in a vacuum centrifuge. 100 µg of peptides of each sample were labeled using 11-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Waltham, MA) following the recent published protocol36. One pooled sample was generated by pooling an aliquot of 25 µg peptides from each sample to serve as a “universal reference”. The reference sample was included as the 11th channel for the two TMT-11 experiments (Supplementary Fig. 1). The TMT-labeled peptides combined from all 11 channels were further fractionated by basic pH reversed-phase LC using a C18 column (250 mm × 2.1 mm, 5 μm particles, Waters, Milford, MA) using an Agilent 1200 HPLC. Ninety-six fractions were collected and concatenated into 24 fractions as previously described37, dried in a vacuum centrifuge, and resuspended in 0.1% formic acid.
For LC-MS/MS analysis, fractionated peptide samples were analyzed using a nanoAquity UPLC system (Waters) coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). LC separations were performed with a custom-packed analytical C18 column (50 cm × 75 µm i.d., 3 µm particle size of Jupiter C18, Phenomenex) with a 120 min gradient. Binary mobile phases comprised of buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in acetonitrile) were used at a flow rate of 300 nl/min. For peptide elution, the percentage of buffer B was increased linearly and a 10 min wash with 95% buffer B and a final 1 min wash with 100% buffer B was also included. Obitrap full MS scans were conducted from 350 to 1800 m/z with a resolution of 60 K and AGC target of 4 × 105 followed by data-dependent higher-energy collisional dissociation (HCD) MS/MS acquisitions at a resolution of 50 K (AGC 1 × 105) and a maximum injection time (IT) of 105 ms for a total cycle time of 2 s. The MS/MS isolation window was set as 0.7 m/z with HCD normalized collision energy of 32. Peptide mode was selected for monoisotopic precursor scan and charge state screening was enabled to reject unassigned 1+, 7+, 8+, and >8+ ions with a dynamic exclusion time of 45 s to discriminate against previously analyzed ions between ± 10 ppm.
For data processing and analysis, the thermo RAW files were first processed with mzRefinery to characterize and correct for any instrument calibration errors, and then with MS-GF+ v2109.08.2638 to match against the UniProt human proteome database (2019.11.05 release; 20,352 entries). A decoy database from the searched fasta files was created by MS-GF+ to estimate the FDR. As searching parameters, the parent ion mass tolerance was set at 20 ppm, 2 missed cleavages were allowed. Cysteine carbamidomethylation (+57.0215) and N-terminal/lysine TMT labeling (229.1629) were searched as static modifications, whereas methionine oxidation (15.9949) was set as variable modification. Spectral-peptide matches were filtered using PepQValue < 0.005 and <7 ppm resulting in maximum FDR of 1%. A minimum of 6 unique peptides per 1000 amino acids of protein length was then required for achieving 1% at the protein level within the full data set. Post-processing of quantitative data of TMT reporter ion intensities was performed using a R package “PlexedPiper” for isobaric quantification [https://github.com/PNNL-Comp-Mass-Spec/PlexedPiper] similar as previously reported39. Briefly, the intensities of all 11 TMT reporter ions were extracted using MASIC software (v 3.0.7235) [https://github.com/PNNL-Comp-Mass-Spec/MASIC/]40. The reporter ion intensities from different scans and different fractions corresponding to the same gene were grouped. Relative protein abundance was calculated as the ratio of abundances between individual sample channels to the reference channel using the summed reporter ion intensities from peptides that could be uniquely mapped to a gene. The relative abundances were log2 transformed and zero-centered for each protein to obtain final relative abundance values. Statistical analyses to test protein abundance changes over time due to either drug effect or progression of T1D were performed using a mixed effects model, where treatment group and timepoint factors were modeled as fixed effect and subjects were modeled as random effect. The significance of the drug treatment was tested using interaction between the group and timepoint effects. Thus, the full model was formulated as protein ~ timepoint:group + timepoint + group + (1|subject). The significance of the timepoint:group was tested using the nested model approach. The test was two-tailed and was performed using lme4 package [doi:10.18637/jss.v067.i01] of R language for statistical computing [https://www.R-project.org/]. Proteins with significant changes in abundance were further analyzed using Enrichr Gene Ontology Biological Process 2021 term enrichment2.
ELISA
Serum CHGA levels were assessed using the Human Chromogranin A ELISA Kit (Epitope Diagnostics, INC., San Diego, CA). Serum IL-21 levels were measured using the Human IL-21 Proquantum Immunoassay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions (limits of quantitation: 0.32–5000 pg/mL).
Quantitative real-time PCR
Total RNA was extracted from PBMCs using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was reverse transcribed to cDNA using the first strand cDNA synthesis kit (Roche) and quantitative real-time PCR (qPCR) was performed on a LightCycler 480 system (Roche) as reported previously41. Relative expression of the proinflammatory T-cell markers CD4, CXCR3, STAT4, CXCR5, and IL21 was measured using the primers listed in Supplementary Table 7. Assessment of markers such as IL21 by qPCR using PBMCs has been successful in the past12,42,43. All results were normalized for GAPDH run as an internal standard and serial template dilutions were performed to confirm comparable target and reference amplification efficiency. The data was then analyzed using the 2ˆ(-ddCT) method as previously described in detail44.
Transcriptomics/RNA sequencing
Human islets from three different donors were obtained from the Integrated Islet Distribution Program (IIDP) and after overnight incubation at 5 mM glucose, 250 islets were handpicked per sample and incubated for 24 h in 25 mM glucose RPMI 1640 medium with or without 100 μM of verapamil prior to RNA extraction using a miRNeasy Mini Kit (Qiagen, Germantown, MD). This concentration of verapamil has been established in early studies for the use in in vitro experiments using beta cells and islets45,46.
RNA sequencing was performed by Exiqon/Qiagen and included preparation of libraries using TruSeq stranded mRNA sample preparation kit (Illumina Inc., San Diego, CA) and single-end sequencing was performed with an average of ~43 million reads obtained per sample.
For data processing and analysis, RNA-sequencing reads were aligned to the H. sapiens reference genome (GRCh38.p7) using STAR (v2.4.2a) with an average ~90% reads uniquely mapped. Alignments were quantified using Salmon (v0.8.2) and differential expression analysis was performed using DESeq2. The DESeq2 model accounted for the experimental design of paired treated and untreated samples from each individual. DESeq2 was used to determine the significance of the differential expression. FDR was calculated using the Benjamini–Hochberg method. Significantly downregulated genes were further analyzed using Enrichr Gene Ontology Biological Process 2021 term enrichment2.
Statistical analysis
All available data were included in the analysis and no data from adequate samples were excluded. Statistician and experimenters were blinded to the study group allocation of samples. Population characteristics of study subjects were summarized as mean and standard errors (SEM) for continuous variables and frequencies for categorical variables. The group comparison of baseline measures was conducted using Chi-square test, Fisher’s exact test, or t-test where appropriate. The normal distribution assumption was checked using Q–Q plots and nonparametric analyses were performed where appropriate. To evaluate the effects of time and group, two-way repeated-measures ANOVA was used. One-way ANOVA or Kruskal–Wallis followed by Dunn’s multiple comparison testing was used to assess the significance between multiple groups. All tests were two-sided. Statistical analyses were performed using SigmaStat 4.0 and SAS 9.4 (Cary, NC).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The proteomics data that support the findings of this study have been deposited in ProteomeXchange with accession: PXD026601.
The MS raw datasets can also be found in the online repositories: Massive.ucsd.edu with accession: MSV000087598.
The transcriptomics data have been deposited in GEO with accession: GSE181328.
Publicly available data sets used can be accessed at:
[https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.33/] (H. sapiens reference genome GRCh38.p7) and [https://www.uniprot.org/proteomes/UP000005640] (UniProt human proteome database (2019.11.05 release).
Remaining data are available within the Article, Supplementary Information, or the Source Data provided with this paper. Source data are provided with this paper.
References
Ovalle, F. et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat. Med. 24, 1108–1112 (2018).
**e, Z. et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 1, e90 (2021).
Palmer, J. P. et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: Report of an ADA workshop, 21-22 October 2001. Diabetes 53, 250–264 (2004).
Greenbaum, C. J. et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite type 1. Diabetes TrialNet Data. Diabetes 61, 2066–2073 (2012).
Greenbaum, C. J. et al. Mixed-meal tolerance test versus glucagon stimulation test for the assessment of beta-cell function in therapeutic trials in type 1 diabetes. Diabetes Care 31, 1966–1971 (2008).
Ebert, A., Konig, J., Frommer, L., Schuppan, D. & Kahaly, G. J. Chromogranin serves as novel biomarker of endocrine and gastric autoimmunity. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgaa288 (2020).
Broedbaek, K. & Hilsted, L. Chromogranin A as biomarker in diabetes. Biomark. Med. 10, 1181–1189 (2016).
Gottlieb, P. A. et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J. Autoimmun. 50, 38–41 (2014).
Stadinski, B. D. et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 11, 225–231 (2010).
He, Z. et al. Elevated serum levels of interleukin 21 are associated with disease severity in patients with psoriasis. Br. J. Dermatol. 167, 191–193 (2012).
Mizutani, H., Tamagawa-Mineoka, R., Nakamura, N., Masuda, K. & Katoh, N. Serum IL-21 levels are elevated in atopic dermatitis patients with acute skin lesions. Allergol. Int. 66, 440–444 (2017).
Kenefeck, R. et al. Follicular helper T cell signature in type 1 diabetes. J. Clin. Invest. 125, 292–303 (2015).
Ferreira, R. C. et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 58, 781–790 (2015).
Shao, F., Zheng, P., Yu, D., Zhou, Z. & Jia, L. Follicular helper T cells in type 1 diabetes. FASEB J. 34, 30–40 (2020).
Baharlou, R. et al. Elevated levels of T-helper 17-associated cytokines in diabetes type I patients: Indicators for following the course of disease. Immunol. Invest. 45, 641–651 (2016).
Citro, A., Campo, F., Dugnani, E. & Piemonti, L. Innate immunity mediated inflammation and beta cell function: Neighbors or enemies? Front. Endocrinol. 11, 606332 (2020).
Strawbridge, R. J. et al. Soluble CD93 is involved in metabolic dysregulation but does not influence carotid intima-media thickness. Diabetes 65, 2888–2899 (2016).
Menger, M. M. et al. Erythropoietin exposure of isolated pancreatic islets accelerates their revascularization after transplantation. Acta Diabetol. https://doi.org/10.1007/s00592-021-01760-4 (2021).
Richardson, S. J. et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 59, 2448–2458 (2016).
Colli, M. L. et al. An integrated multi-omics approach identifies the landscape of interferon-alpha-mediated responses of human pancreatic beta cells. Nat. Commun. 11, 2584 (2020).
Russell, M. A. et al. HLA class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet beta-cells from donors with type 1. Diabetes Diabetes 68, 988–1001 (2019).
Chen, K. et al. Overexpression of Insig-1 protects beta cell against glucolipotoxicity via SREBP-1c. J. Biomed. Sci. 18, 57 (2011).
Cogger, K. F. et al. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nat. Commun. 8, 331 (2017).
Zhang, T. et al. Common variants in NUS1 and GP2 genes contributed to the risk of gestational diabetes mellitus. Front. Endocrinol. 12, 685524 (2021).
Werner, L. et al. Identification of pancreatic glycoprotein 2 as an endogenous immunomodulator of innate and adaptive immune responses. J. Immunol. 189, 2774–2783 (2012).
Xu, G., Chen, J., **g, G. & Shalev, A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 61, 848–856 (2012).
Chen, J., Saxena, G., Mungrue, I. N., Lusis, A. J. & Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta cell apoptosis. Diabetes 57, 938–944 (2008).
Chen, J., Fontes, G., Saxena, G., Poitout, V. & Shalev, A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated beta-cell death. Diabetes 59, 440–447 (2010).
Chen, J. et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta cell mass and protects against diabetes. FASEB J. 22, 3581–3594 (2008).
Kudo, M. et al. Oxidative stress induced interleukin-32 mRNA expression in human bronchial epithelial cells. Respir. Res. 13, 19 (2012).
Kallionpaa, H. et al. Early detection of peripheral blood cell signature in children develo** beta-cell autoimmunity at a young age. Diabetes 68, 2024–2034 (2019).
Poma, A. M. et al. Immune transcriptome of cells infected with enterovirus strains obtained from cases of type 1 diabetes. Microorganisms https://doi.org/10.3390/microorganisms8071031 (2020).
Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. The diabetes control and complications trial research group. Ann. Intern. Med. 128, 517–523 (1998).
Keshishian, H. et al. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 12, 1683–1701 (2017).
Liu, T. et al. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol. Cell Proteom. 5, 2167–2174 (2006).
Zecha, J. et al. TMT labeling for the masses: A robust and cost-efficient, in-solution labeling approach. Mol. Cell Proteom. 18, 1468–1478 (2019).
Wang, Y. et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 (2011).
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277 (2014).
Zhang, H. et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 (2016).
Monroe, M. E., Shaw, J. L., Daly, D. S., Adkins, J. N. & Smith, R. D. MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217 (2008).
Jo, S., Xu, G., **g, G., Chen, J. & Shalev, A. Human glucagon expression is under the control of miR-320a. Endocrinology https://doi.org/10.1210/endocr/bqaa238 (2021).
Publicover, J. et al. IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B. J. Clin. Invest. 121, 1154–1162 (2011).
Xu, X. et al. Inhibition of increased circulating Tfh cell by anti-CD20 monoclonal antibody in patients with type 1 diabetes. PLoS One 8, e79858 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Kennedy, H. J., Rafiq, I., Pouli, A. E. & Rutter, G. A. Glucose enhances insulin promoter activity in MIN6 beta-cells independently of changes in intracellular Ca2+ concentration and insulin secretion. Biochem. J. 342, 275–280 (1999).
Simonsson, E., Karlsson, S. & Ahren, B. Ca2+-independent phospholipase A2 contributes to the insulinotropic action of cholecystokinin-8 in rat islets: Dissociation from the mechanism of carbachol. Diabetes 47, 1436–1443 (1998).
Acknowledgements
This work was supported by grants from the National Institutes of Health to A.S. (R01DK078752 and Human Islet Research Network U01DK120379) and to W.J.Q. (DP3DK110844 and R01DK122160) and ADA Pathway Award to P.S. (1-16-ACE-47). The UAB Human Physiology Core was supported by NIH award P30DK079626. Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) (RRID:SCR_014387) at City of Hope, NIH grant UC4DK098085. Mass spectrometry proteomics experiments were performed in the Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, a national scientific user facility sponsored by the DOE under Contract DE-AC05-76RL0 1830.
Author information
Authors and Affiliations
Contributions
G.X. conducted the molecular analyses and generated the figures, G.X., L.A.T., and T.B.G. helped with sample processing and, F.O. and T.G. were responsible for the MMTTs and sample and data collection. J.C. helped with the human islet processing, H.M.T. provided immunological expertise and P.L. helped with the statistical analysis. M.K. and P.S. were responsible for the RNA sequencing and A.A.S., A.C.S., T.T.L., V.A.P., and W.J.Q. conducted the mass spectrometry studies and analyses. A.S. designed the studies, analyzed the results, and wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Peter Rossing and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, G., Grimes, T.D., Grayson, T.B. et al. Exploratory study reveals far reaching systemic and cellular effects of verapamil treatment in subjects with type 1 diabetes. Nat Commun 13, 1159 (2022). https://doi.org/10.1038/s41467-022-28826-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28826-3
- Springer Nature Limited