
Abstract
The upcoming flu season in the Northern Hemisphere merging with the current COVID-19 pandemic raises a potentially severe threat to public health. Through experimental coinfection with influenza A virus (IAV) and either pseudotyped or live SARS-CoV-2 virus, we found that IAV preinfection significantly promoted the infectivity of SARS-CoV-2 in a broad range of cell types. Remarkably, in vivo, increased SARS-CoV-2 viral load and more severe lung damage were observed in mice coinfected with IAV. Moreover, such enhancement of SARS-CoV-2 infectivity was not observed with several other respiratory viruses, likely due to a unique feature of IAV to elevate ACE2 expression. This study illustrates that IAV has a unique ability to aggravate SARS-CoV-2 infection, and thus, prevention of IAV infection is of great significance during the COVID-19 pandemic.
Similar content being viewed by others


Introduction
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) at the end of 2019 has led to a worldwide pandemic. Until 13 January 2021, there have been more than 90 million confirmed infection cases and 1.9 million deaths globally (https://covid19.who.int/). The ending time and the final severity of the current COVID-19 pandemic wave are still uncertain. Meanwhile, the influenza season is merging with the current pandemic, potentially bringing more challenges and posing a larger threat to public health. There are many debates on whether seasonal flu will impact the severity of the COVID-19 pandemic and whether influenza vaccination is necessary for the coming winter. However, no experimental evidence is available concerning IAV and SARS-CoV-2 coinfection.
It is well known that the disease symptoms of SARS-CoV-2 and IAV infection symptoms are quite similar, including fever, cough, pneumonia, and acute respiratory distress syndrome.1,2 Moreover, both SARS-CoV-2 and IAV are airborne transmitted pathogens that infect the same human tissues, namely, the respiratory tract and nasal, bronchial, and alveolar epithelial cultures.3,4 In addition, alveolar type II cells (AT2 pneumocytes) appear to be preferentially infected by SARS-CoV-2 and are also the primary site of IAV replication.5,6 Therefore, overlap of the COVID-19 pandemic and seasonal influenza might place a large population under high risk for concurrent infection with these two viruses.7
Unfortunately, during the last winter flu season in the southern hemisphere, little epidemiological evidence was collected regarding the interaction between COVID-19 and flu, likely due to a low IAV infection rate resulting from social distancing.8 One case report showed that three out of four SARS-CoV-2 and IAV coinfected patients rapidly develop respiratory deterioration.9 In contrast, another study only reported mild symptoms in limited coinfection outpatients.10 A retrospective study found that the coinfection rate of SARS‐CoV‐2 and influenza virus was as high as 57.3% (among which 49.8% was coinfected with IAV) in a single‐centered study of 307 COVID-19 patients during the outbreak period in Wuhan.11 Thus, the high coinfection rate and the unpredictable clinical outcomes pose great concerns when facing the threat of both viruses.
In this study, we tested whether IAV infection could affect the subsequent SARS-CoV-2 infection in both cultured cells and mice. Our results demonstrate that preinfection with IAV strongly enhances the infectivity of SARS-CoV-2 by boosting viral entry into cells and elevating the viral load, leading to more severe lung damage in infected mice. These data suggest a clear auxo-action of IAV on SARS-CoV-2 infection, which underscores the great risk of influenza virus and SARS-CoV-2 coinfection to public health.
Results
IAV promotes SARS-CoV-2 virus infectivity
To study the interaction between IAV and SARS-CoV-2, A549 (a hypotriploid alveolar basal epithelial cell line) cells that are susceptible to IAV infection but usually do not support SARS-CoV-2 infection were applied to test whether IAV preinfection would modulate the infectivity of SARS-CoV-2. Pseudotyped VSV luciferase-reporter particles bearing SARS-CoV-2 spike protein (pseudo-SARS-CoV-2) were used to visualize the viral entry.28 Overall, the fact that the IAV segment might promote SARS-CoV-2 infection further confirms a unique positive interaction between IAV and SARS-CoV-2.
Importantly, the enhancement phenotype in IAV and SARS-CoV-2 coinfection was independent of IFN signaling and was not observed when cells were coinfected with inactivated IAV (data not shown). Therefore, influenza vaccination (especially inactivated influenza vaccine) to inhibit influenza infection should be recommended to people with a high risk of coinfection. Our findings also remind that surveillance of coinfection is encouraged in the coming winter. Moreover, social distancing and mask-wearing are beneficial to protect people from the transmission of either or both viruses.
Materials and methods
Cells and viruses
The 293 T, A549, Huh-7, MDCK, Vero E6, WI-38, WI-38 VA-13, and BEAS-2B cells were obtained from ATCC and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS). The Calu-3 cells (ATCC) were maintained in DMEM supplemented with 20% FBS. NCI-H292 cells (ATCC) were maintained in RPMI-1640 (Gibco) supplemented with 20% FBS. Normal human bronchial epithelial cells (NHBE) cells (ATCC) were maintained in airway epithelial cell basal medium (ATCC PCS300030) supplemented with a Bronchial/Tracheal Epithelial Cell Growth Kit (ATCC PCS300040). All cells were incubated at 37 °C in 5% CO2.
The A/WSN/33 virus was generated by reverse genetics as previously described.29 H1N1 (A/Sichuan/01/2009) and H3N2 (A/Donghu/312/2006) were kindly provided by the Influenza Center in China CDC. HRV3 and HRSV were purchased from ATCC and stocked accordingly. HPIV was obtained from Professor Mingzhou Chen, Wuhan University. The SARS-CoV-2 live virus (strain IVCAS 6.7512) was provided by the National Virus Resource, Wuhan Institute of Virology, Chinese Academy of Sciences.
Plasmids and transfection
The plasmid encoding SARS-CoV-2-S-Δ18 was a gift from Professor Ningshao **a, **amen University. The eight WSN viral segments in the pHW2000 plasmid were kindly provided by Professor Hans Klenk, Marburg University. The DNA transfection reagent Fugene HD was purchased from Promega, and the transfection was performed according to manuscript procedures.
Pseudotype virus production
Pseudotyped VSV-ΔG viruses expressing either a luciferase reporter or mCherry reporter were provided by Professor Ningshao **a, **amen University. To produce pseudotyped VSV-ΔG-Luc/mCherry bearing SARS-CoV-2 spike protein (pseudo-SARS-CoV-2), Vero E6 cells were seeded in 10-cm dishes and transfected simultaneously with 15 μg SARS-CoV-2-S-Δ18 plasmid using Lipofectamine 3000 (Thermo). Forty-eight hours posttransfection, 150 µL pseudotyped VSV-ΔG bearing VSV-G protein was used to infect Vero E6 cells. Cell supernatants were collected after another 24 h, clearing of cell debris by centrifugation at 3000 rpm for 6 min, aliquoted, and stored at −80 °C.
Luciferase-based cell entry assay
Target cells were seeded in 48-well plates and inoculated in triplicate with equivalent volumes of pseudotyped virus stocks at a 1:5 dilution in DMEM (3% FBS) with or without IAV preinfection. At 24 h post-pseudotype infection, luciferase activities were measured with a Luciferase Assay System (Promega E4550).
Virus infection and IFN treatment
For IAV infection, cells were washed with PBS and then incubated with viruses at different MOIs (from 0.01 to 1) in infection medium (DMEM, supplemented with 2% FBS, 1% penicillin/streptomycin) at 37 °C in 5% CO2.
For SARS-CoV-2 infection, cells were incubated with SARS-CoV-2 live virus at an MOI of 0.01 in infection medium (DMEM, 1% penicillin/streptomycin) and incubated at 37 °C in 5% CO2 for 1 h with or without 12 h IAV preinfection (MOI 0.1). Cells were then washed with PBS two times and incubated in culture medium (DMEM, supplemented with 5% FBS and 1% penicillin/streptomycin) at 37 °C in 5% CO2 for 48 h.
For HRV3, HPIV or HRSV infection, cells were washed with PBS and then incubated with the indicated viruses in infection medium (DMEM, supplemented with 3% FBS, 1% penicillin/streptomycin) and incubated at 37 °C in 5% CO2 for 12 h.
For IFN treatment, recombinant human IFNα2a (Beyotime, P5646) and IFNγ (Beyotime, P5664) were dissolved in 0.1% BSA, diluted in DMEM with 10% FBS, and then incubated with cells for 12 h at the indicated doses.
Real-time reverse-transcriptase–polymerase chain reaction
The mRNA levels of the indicated genes were quantified via quantitative PCR with reverse transcription (qRT-PCR). Purified RNAs extracted with TRIzol (Invitrogen™,15596018) were subjected to reverse transcription using oligo dT or random primer (using Takara cat#RR037A Kit), and then, the corresponding cDNAs were quantified using Hieff qPCR SYBR Green Master Mix (Yeason). Thermal cycling was performed in a 384-well reaction plate (ThermoFisher, 4343814). Quantification of IAV replication was measured by SYBR Green qRT-PCR with primers targeting NP vRNA, and the IAV expression was measured by SYBR Green qRT-PCR with primers targeting NP mRNA. Quantification of the propagation for other respiratory viruses was measured by SYBR Green qRT-PCR with primers targeting the HRSV M gene, the HPIV M gene, and the HRV3 5′UTR (position 456–569), respectively. Host receptor and cofactor genes were measured using gene-specific primers (Supplementary information, Table S1). All mRNA levels were normalized to the β-actin level in the same cell.
The relative number of SARS-CoV-2 viral genome copies was determined using a TaqMan RT-PCR Kit (Yeason). To accurately quantify the absolute number of SARS-CoV-2 genomes, a standard curve was prepared by measuring the SARS-CoV-2 N gene constructed in the pCMV-N plasmid. All SARS-CoV-2 genome copy numbers were normalized to GAPDH expression in the same cell.
All the primers and Tagman probes used in this study were listed in Supplementary information, Table S1.
Western blot analysis
For western blot analysis, cells were lysed in RIPA buffer on ice for 30 min, separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to western blot analysis. For mouse experiments, half of the lung tissue from each mouse was homogenized in PBS, followed by boiling in SDS lysis buffer (GE) at 100 °C for 30 min. Rabbit monoclonal antibody against ACE2 (Abclonal, A4612, 1:1000), mouse monoclonal antibody against SARS-CoV Nucleoprotein (Sino Biological, 40143-MM05, 1:1000), and anti-actin antibody (Abclonal, 1:1000) were purchased commercially. The anti-influenza virus-NP antibody was kindly provided by Professor Ningshao **a. Peroxidase-conjugated secondary antibodies (Antgene, 1: 5000) were applied accordingly, followed by image development with a Chemiluminescent HRP Substrate Kit (Millipore Corporation).
Immunofluorescence
A549 cells were fixed and incubated with primary antibodies. The primary antibodies used in this study were rabbit polyclonal antibody against ACE2 for immunofluorescence (Sino Biological, 10108-T26) and anti-influenza virus-NP antibody (kindly provided by Professor Ningshao **a). Alexa Fluor dye-conjugated secondary antibodies (Alexa Fluor R488, Invitrogen; Alexa Fluor M555, Invitrogen) and DAPI (Beyotime, C1002) were administered afterward according to standard protocols. Cell imaging was performed on a Leica TCS SP8 confocal laser scanning microscope (Leica).
ACE2 knockdown cells
Two sgRNAs targeting the hACE2 gene were designed under the protocol in http://chopchop.cbu.uib.no (sgRNA sequence of ACE2 were shown in Supplementary information, Table S1) and commercially synthesized to clone into the lenti-Cas9-blast vector (kindly provided by Professor Hongbing Shu). The control sgRNA lentivirus construct was also provided by Professor Hongbing Shu. In brief, A549 cells were plated in 6-well plates and transduced with lentivirus encoding the CRISPR-Cas9 system, including either ACE2 sgRNA or control sgRNA. The cell mixtures were selected with blasticidin for one week to obtain ACE2 knockdown cells. The gene knockdown efficiencies were confirmed by measuring the ACE2 mRNA level through qRT-PCR analysis.
Mice
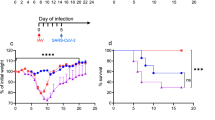
K18-hACE2 transgenic mice, which express human ACE2 driven by the human epithelial cell cytokeratin-18 (K18) promoter used as an infection model for both SARS-CoV and SARS-CoV-2,30,31,32 were purchased from Gempharmatech and housed in ABSL-3 pathogen-free facilities under 12-h light-dark cycles with access to food and water. All animal experiments were approved by the Animal Care and Use Committee of Wuhan University. Mice were male, age-matched, and grouped for SARS-CoV-2 infection or IAV and SARS-CoV-2 coinfection. On day 0, mice were intranasally infected with PBS or 2000 PFU of WSN, and then, both groups were intranasally infected with 3 × 105 PFU of SARS-CoV-2 on day 2. Another two days later, mice were sacrificed to determine viral loads and for histological assays.
Histology analysis
Lung tissue from infected mice was dissected on day 2 post-SARS-CoV-2 infection, fixed, and stained using a standard Hematoxylin-Eosin staining (H&E) staining procedure. The slides were scanned and analyzed by the Wuhan Sci-Meds company. Representative images from three mice in each group are shown.
Statistical analysis
If not indicated otherwise, Student’s t-test was used for two-group comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 were considered significant. Unless otherwise noted, error bars indicate mean values and standard deviations of at least three biological experiments.
References
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Van Riel, D. et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am. J. Pathol. 171, 1215–1223 (2007).
St. John, A. L. & Rathore, A. P. S. Early insights into immune responses during COVID-19. J. Immunol. 205, 555 (2020).
Traylor, Z. P., Aeffner, F. & Davis, I. C. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza and other respiratory. Viruses 7, 472–479 (2013).
Hou, Y. et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182, 429–446 (2020).
Belongia, E. & Osterholm, M. COVID-19 and flu, a perfect storm. Science 368, 1163 (2020).
Olsen, S. et al. Decreased influenza activity during the COVID-19 Pandemic - United States, Australia, Chile, and South Africa, 2020. MMWR Morb. Mortal Wkly. Rep. 69, 1305–1309 (2020).
Cuadrado-Payán, E. et al. SARS-CoV-2 and influenza virus co-infection. Lancet 395, e84 (2020).
Zheng, X. et al. Co-infection of SARS-CoV-2 and influenza virus in early stage of the COVID-19 epidemic in Wuhan, China. J. Infect. 81, e128–e129 (2020).
Yue, H. et al. The epidemiology and clinical characteristics of co-infection of SARS-CoV-2 and influenza viruses in patients during COVID-19 outbreak. J. Med. Virology 92, 2870–2873 (2020).
**ong, H.L. et al. Robust neutralization assay based on SARS-CoV-2 S-protein-bearing vesicular stomatitis virus (VSV) pseudovirus and ACE2-overexpressing BHK21 cells. Emerg. Microbes Infect. 9, 2105–2113 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904.e9 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e278 (2020).
Wu, C. et al. Furin, a potential therapeutic target for COVID-19. iScience 23, 101642 (2020).
Grimm, C. & Tang, R. Could an endo-lysosomal ion channel be the achilles heel of SARS-CoV2? Cell Calcium 88, 102212 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e286 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e1019 (2020).
Priyamvada, L. et al. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc. Natl. Acad. Sci. USA 113, 7852 (2016).
Mateus, J. et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370, 89–94 (2020).
Shi, G. et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 0, e106501 (2020).
Vanderheiden, A. et al. Type I and Type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J. Virol. 94, e00985–00920 (2020).
Lokugamage, K. G. et al. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 94, e01410–e01420 (2020).
Wise, H. M. et al. A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 83, 8021–8031 (2009).
Wang, R. et al. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy https://doi.org/10.1080/15548627.2020.1725375 (2020).
Wise, H. et al. Overlap** signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res. 39, 7775–7790 (2011).
Han, Q. et al. Sumoylation of influenza A virus nucleoprotein is essential for intracellular trafficking and virus growth. J. Virology 88, 9379–9390 (2014).
McCray, P. B. Jr et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virology 81, 813–821 (2007).
Zheng, J. et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589, 603–607 (2021).
Winkler, E. S. et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 21, 1327–1335 (2020).
Acknowledgements
This work was supported in part by the National Key R&D Program (2018FYA0900801 to K.X. and 2016YFA0502103 to K.L.), the National Natural Science Foundation of China (grants 31922004 and 81772202 to K.X.), Application & Frontier Research Program of the Wuhan Government (2019020701011463 to K.X.), and Hubei Innovation Team Foundation (2020CFA015 to K.X. and K.L.).
Author information
Authors and Affiliations
Contributions
K.X. and K.L. conceived the project and designed the experiments. L.B., J.D., M.G., X.W., Z.H., Z.Z., and YC. Z. coordinated the live SARS-CoV-2 study and performed animal infection experiments. YL. Z. and S. L. conducted pseudotyped virus infection experiments, IFN treatment experiments, and data analysis. L.B. and J.D. evaluated the immunofluorescence, histopathological and immunohistochemical studies. X.L. performed HRV3, HPIV, and HRSV infection experiments. YL. Z and X. L. generated the mutant virus and performed the related tests. L.B., S.L., J.D., and X.L. repeated the key experiments in infected cells. X.S., Q.L., D.N., M.X., K.S., J.Y., W.Z., Z.T., M.T., Y.Z., C.S., M.D., L.Z., Y.C., and H.Y. provided technical support and materials. L. D. constructed ACE2 knockout cells and conducted related analysis. K.X., K.L., S.L., and YL. Z. wrote the manuscript with input from all the other authors. For their research spirit and courage, we also thank our group members of the SARS-CoV-2 working group in the State Key Laboratory of Virology, Wuhan University, who are working closely together during this new virus pandemic. We are grateful to Taikang Insurance Group Co., Ltd; Bei**g Taikang Yicai Foundation; and Special Fund for COVID-19 Research of Wuhan University for their great supports of this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bai, L., Zhao, Y., Dong, J. et al. Coinfection with influenza A virus enhances SARS-CoV-2 infectivity. Cell Res 31, 395–403 (2021). https://doi.org/10.1038/s41422-021-00473-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-021-00473-1
- Springer Nature Singapore Pte Ltd.
This article is cited by
-
Advances in nanobiosensors during the COVID-19 pandemic and future perspectives for the post-COVID era
Nano Convergence (2024)
-
Immunogenicity and protective efficacy of a co-formulated two-in-one inactivated whole virus particle COVID-19/influenza vaccine
Scientific Reports (2024)
-
An RNA-hydrolyzing recombinant minibody prevents both influenza A virus and coronavirus in co-infection models
Scientific Reports (2024)
-
Influenza a virus regulates interferon signaling and its associated genes; MxA and STAT3 by cellular miR-141 to ensure viral replication
Virology Journal (2023)
-
Epidemiology and molecular characteristics of respiratory syncytial virus (RSV) among italian community-dwelling adults, 2021/22 season
BMC Infectious Diseases (2023)