
Abstract
Chronic obstructive pulmonary disease (COPD) is a risk factor for lung cancer development. COPD induces activation of hypoxia-induced signaling, causing remodeling of surrounding microenvironmental cells also modulating the release and cargo of their extracellular vesicles (EVs). We aimed to evaluate the potential role of circulating EVs from COPD subjects in lung cancer onset. Plasma-EVs were isolated by ultracentrifugation from heavy smoker volunteers with (COPD-EVs) or without (heavy smoker-EVs, HS-EV) COPD and characterized following MISEV guidelines. Immortalized human bronchial epithelial cells (CDK4, hTERT-HBEC3-KT), genetically modified with different oncogenic alterations commonly found in lung cancer (sh-p53, KRASV12), were used to test plasma-EVs pro-tumorigenic activity in vitro. COPD-EVs mainly derived from immune and endothelial cells. COPD-EVs selectively increased the subset of CD133+CXCR4+ metastasis initiating cells (MICs) in HBEC-sh-p53-KRASV12high cells and stimulated 3D growth, migration/invasion, and acquisition of mesenchymal traits. These effects were not observed in HBEC cells bearing single oncogenic mutation (sh-p53 or KRASV12). Mechanistically, hypoxia-inducible factor 1-alpha (HIF-1α) transferred from COPD-EVs triggers CXCR4 pathway activation that in turn mediates MICs expansion and acquisition of pro-tumorigenic effects. Indeed, HIF-1α inhibition or CXCR4 silencing prevented the acquisition of malignant traits induced by COPD-EVs alone. Hypoxia recapitulates the effects observed with COPD-EVs in HBEC-sh-p53-KRASV12high cells. Notably, higher levels of HIF-1α were observed in EVs from COPD subjects who subsequently developed cancer compared to those who remained cancer-free. Our findings support a role of COPD-EVs to promote the expansion of MICs in premalignant epithelial cells through HIF-1α-CXCR4 axis activation thereby potentially sustaining lung cancer progression.
Similar content being viewed by others

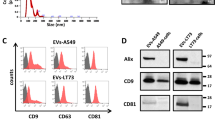

Introduction
Cigarette smoking can be considered the main risk factor for the development of chronic obstructive pulmonary disease (COPD) and for the onset of lung cancer [1]. However, only 20% of smokers develop COPD [2], whereas the incidence of lung cancer is higher in subjects with COPD [3]. Since COPD and lung cancer display the activation of some common pathways related to inflammation or growth factors [4], it is likely that the presence of COPD could be a potent driver for lung cancer progression. In NSCLC, we and other shown that CD133 represents a robust marker to identify cells endowed with stem like properties and cancer initiating potential (cancer-initiating cells-CICs) [5,6,7]. It is now well-recognized that CICs represent an heterogeneous population comprised of cells deputed to specific function, endowed with different properties and characterized by specific markers [8]. In lung cancer for instance different markers have been described to possibly identify CIC subset, suggesting a great phenotypic heterogeneity of such population [9,10,Full size image
Based on these preliminary results, we focused our attention on HBEC-KRASV12high in which a more striking effect in the acquisition of a malignant phenotype was observed after COPD-EVs treatment. We further observed that the treatment with COPD-EVs significantly increased the percentage of wound closure compared to HS-EVs (% wound closure: COPD-EVs= 75.8%, SEM = 5.1%; HS-EVs= 58.2%, SEM = 4.8%; NT = 60.6%, SEM = 3.1%. p = 0.0451 vs. HS-EVs and p = 0.042 vs. NT) (Fig. 3G and Supplementary Fig. S3). Instead, proliferation assay showed that COPD-EVs did not affect the growth rate of HBEC-KRASV12high, cultured as monolayer (Fig. 3H); however, COPD-EVs but not HS-EVs fostered the 3D-colony forming ability of HBEC-KRASV12high, providing and indirect indication on the pro-tumorigenic features of COPD-EVs treated cells (Fig. 3I). Overall, these results suggest that COPD-EVs promote the increase of CD133+ cancer-initiating cells, in particular through the expansion of CD133+CXCR4+ MICs subset, along with the acquisition of pro-tumorigenic and invasive properties in HBEC-KRASV12high cells.
HIF-1α is transferred from COPD-EVs to HBEC-KRASV12high
To investigate the factors causing the increase of MICs subset and the acquisition of pro-tumorigenic properties induced by COPD-EVs we focused on SDF-1, the cytokine that activates CXCR4 downstream signaling pathways, also known to regulate CICs phenotype [30]. Treatment of HBEC-KRASV12high cells with neutralizing antibody against SDF-1 did not abrogate the modulation of CICs cells induced by COPD-EVs, indicating that SDF-1-CXCR4 activation axis was not central in COPD-EVs induced expansion of CICs (Fig. 4A). Since it has been demonstrated that COPD causes a hypoxic lung microenvironment, we decided to analyze inside EVs the amount of the transcription factor HIF-1α, the master regulator of the cellular response to hypoxia. HIF-1α level, evaluated via ELISA, was significantly lower in HS-EVs (2.1 pg/ml, SEM = 1.5 pg/ml) than in COPD-EVs (7.5 pg/ml, SEM = 2.0 pg/ml) (*p = 0.03) (Fig. 4B). The transfer of HIF-1α into recipient cells via EVs was evaluated by treating HBEC-KRASV12high with PKH-labeled-EVs. Intracellular level of HIF-1α in PKH-fluorescent positive cells treated with COPD-EVs was 2.2-fold increased compared to the HS-EV treatment (p < 0.01) (Fig. 4C). To further elucidate whether the increase of HIF-1α in recipient cells was actually due to EVs protein shuttling or to the induction of endogenous transcript, levels of HIF-1α mRNA were evaluated after EVs treatment of HBEC-KRASV12high. No differences in HIF-1α transcription were observed between the two groups (Fig. 4D), indicating that HIF-1α was not induced into recipient cells but shuttled by EVs. Moreover, the transfer of HIF-1α from COPD-EVs induced the transcription of known downstream targets such as VEGF and CAIX in recipient cells (Fig. 4D). To assess the role of HIF-1α in the induction of MICs phenotype caused by COPD-EVs, we exploited a selective HIF-1α inhibitor, PX-478, before EVs treatment. HIF-1α inhibition completely abrogates the expansion of HBEC-KRASV12high CD133+ CICs induced by COPD-EVs, especially acting on CD133+CXCR4+ MICs (Fig. 4E). Coherently with effects on tumorigenic CICs subset, the blockade of HIF-1α transfer after COPD-EVs treatment reduced the number of 3D-colonies (Fig. 4F). Furthermore, the inhibition of HIF-1α reversed the pro-migratory effect, towards both SDF-1 and EGF chemoattractants, induced by COPD-EVs (Fig. 4G).

A Analysis of CD133 modulation in HBEC-KRASV12high treated with EVs in the presence of SDF-1 neutralizing antibody (n = 3). B Quantification (pg/µl) of HIF-1α carried by COPD or HS-EVs via ELISA (n = 10). C On the left, representative dot plots show the higher positivity for HIF-1α in HBEC-KRASV12high treated with COPD-EVs compared to HS-EVs. On the right, the bar chart shows the percentage of HIF-1α in PKH26-labeled EV-treated HBEC-KRASV12high using flow cytometry (n = 7). D mRNA expression level of HIF-1α, VEGF and CAIX in EV-treated cells (n = 4). E Flow cytometry analysis of CD133+ and CD133+CXCR4+ cells after concomitant HIF-1α inhibition and EVs administration in HBEC-KRASV12high (n = 5). F Quantification of colonies constituted by HBEC-KRASV12high cells treated with 15 µg of COPD- or HS-EVs and concomitant HIF-1α inhibition in 3D culture condition. Untreated cells (NT) were used as a negative control. G Evaluation of migratory capabilities of HBEC-KRASV12high towards EGF and SDF-1 gradients after the treatment with HS- and COPD-EVs in the presence or not of HIF-1α inhibitor (n = 3). Data are expressed as mean and SEM.
HIF-1α shuttle induces CXCR4 modulation and CD133+ stem cells
Since HIF-1α controls directly CXCR4 expression that in turn modulates CICs phenotype, we investigated its possible involvement as mediator of COPD-EVs induced-CICs effect. We confirmed that CXCR4 transcription was significantly increased in HBEC-KRASV12high cells after COPD-EVs treatment and that the inhibition of HIF-1α completely abrogated this effect (Fig. 5A). Notably, no CXCR4 modulation was observed after HS-EVs administration. Furthermore, CXCR4 silencing in HBEC-KRASV12high recipient cells completely prevented the increase of CD133+ and CD133+CXCR4+ subsets caused by COPD-EVs (Fig. 5B). Of note, the blockade of CXCR4 in HBEC-KRASV12high reverted the capacity of COPD-EVs to promote migration (Fig. 5C) and invasion (Fig. 5D) of these cells. Furthermore, the number of 3D colonies induced by COPD-EVs was reduced after CXCR4 silencing in recipient cells (Fig. 5E).

A The increase of mRNA of CXCR4 in HBEC-KRASV12high induced by COPD-EVs was abrogated by the use of HIF-1α inhibitor. B The silencing of CXCR4 reversed the modulation of CD133+ CICs and CD133+CXCR4+ MICs in COPD-EVs treated. In all experiments, untreated cells were used as control. C The silencing of CXCR4 in HBEC-KRASV12high cells reversed the migratory capability gained by cells treated with COPD-EVs. D Pro-invasive properties given by the treatment with COPD-EVs to HBEC-KRASV12high cells were abolished by the silencing of CXCR4. E Colonies formation induced by COPD-EVs treatment in HBEC-KRASV12high cells was inhibited by CXCR4 silencing. *p < 0.05. Data are expressed as mean and SEM.
All together these data support the crucial role of CXCR4 as a mediator of hypoxia-induced modulation of MIC phenotype.
Hypoxia plays a key role in MIC induction and in cancer development
To further demonstrate the central role of hypoxia in the regulation of MIC phenotype, HBEC-1, HBEC-3, HBEC-5, HBEC-6, and HBEC-KRASV12high were incubated for 72 h at 2% oxygen and the modulation of CD133+CXCR4+ was evaluated. Hypoxia induced a 4-fold increase of MIC cells only in HBEC-KRAS V12high cells (*p = <0.05) unlike other cell lines tested (Fig. 6A). Once again, the HIF-1α blocking (Fig. 6B) or the CXCR4 silencing (Fig. 6C) prevented the expansion of MICs in HBEC-KRASV12high in hypoxic conditions. Finally, we demonstrated that hypoxia promoted migratory and invasive capacities of HBEC-KRASV12high that are abrogated by the inhibition of HIF-1α or CXCR4 in recipient cells towards an EGF gradient (Fig. 6D, E and Supplementary Fig. S4). These results corroborate the observation that the cellular response to hypoxia is central in promoting MIC expansion but only in transformed epithelial cells accumulating multiple oncogenic hits, similar to what we previously observed after COPD-EVs treatment.

A Relative amount of CD133+CXCR4+ MICs in HBEC-1, HBEC-3, HBEC-5, HBEC-6, and HBEC-KRASV12high in normoxic or hypoxic condition (n = 3). B In hypoxic condition, the treatment with anti-HIF-1α of HBEC-KRASV12high inhibits the expansion of CD133+CXCR4+ MICs compared to untreated cells cultured in hypoxia (n = 4). C) MICs expansion in HBEC-KRASV12high was abrogated by inducing the silencing of CXCR4 (n = 4). D Migration assay towards an EGF gradient using HBEC-KRASV12high pre-treated with HIF-1α inhibitor or CXCR4 siRNA and cultured 48 h in hypoxic condition prior to perform the assay. E Invasion assay towards an EGF gradient using HBEC-KRASV12high pre-treated with HIF-1α inhibitor or CXCR4 siRNA and cultured 48 h in hypoxic condition prior to seed cells for the assay. F Evaluation of endothelial markers by using MACSPlex kit on EVs released by endothelial cells cultured in both normoxic and hypoxic conditions (n = 3). G Flow cytometry analysis on HBEC-KRASV12high to evaluate the possible expansion of CIC and MIC subsets following the treatment with END-EVs Normoxia and END-EVs Hypoxia. H ELISA performed to evaluate the level of HIF-1α within EVs isolated from COPD volunteers that subsequently developed (n = 15) or not (n = 13) lung cancer. *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as mean and SEM.
Our MACSPlex analysis in Fig. 1E indicated a significant enrichment of endothelial markers in circulating EVs obtained from COPD subjects, suggesting their endothelial cell origin. To assess whether hypoxic environment occurring in COPD subjects promotes EVs release by endothelial cells, we cultured endothelial cells in hypoxic and standard oxygen culture conditions and we next recovered and analyzed EVs via MACSPlex. Hypoxia induced an increase in endothelial markers on the surface of EVs compared to normal condition (Fig. 6F), confirming that hypoxia boosted the release of EVs from endothelial cells.
We verified that EVs released by endothelial cells, maintained both in normoxic (END-EVs Normoxia) and in hypoxic conditions (END-EVs Hypoxia), have a similar ability to increase the total number of CD133+ cells in HBEC-KRASV12high recipient cells, but EVs released by endothelial cells under hypoxia can specifically expand the pool of CD133+CXCR4+ cells, suggesting their unique ability to activate CXCR4 pathway (Fig. 6G).
Finally, to confirm the involvement of hypoxia in cancer development, we analyzed the level of HIF-1α inside EVs from COPD volunteers, who subsequently developed or not lung cancer. We found significant higher levels of HIF-1α (125.8 pg/ml, SEM = 48.66 pg/ml) in EVs from COPD subjects who subsequently developed cancer (i.e. before CT scan detection) than COPD subjects who remained cancer free for at least three years (12.34 pg/ml, SEM = 2.835 pg/ml) (*p = 0.04) (Fig. 6H).
Taken together, these results highlight the strong involvement of COPD-derived hypoxia in inducing the release of EVs by endothelial cells and the potential role of HIF-1α within EVs in fostering lung cancer development that may be exploited as biomarker of lung cancer risk.
