
Abstract
Peptidylarginine deiminase (PADI) 2 catalyzes the post-translational conversion of peptidyl-arginine to peptidyl-citrulline in a process called citrullination. However, the precise functions of PADI2 in bone formation and homeostasis remain unknown. In this study, our objective was to elucidate the function and regulatory mechanisms of PADI2 in bone formation employing global and osteoblast-specific Padi2 knockout mice. Our findings demonstrate that Padi2 deficiency leads to the loss of bone mass and results in a cleidocranial dysplasia (CCD) phenotype with delayed calvarial ossification and clavicular hypoplasia, due to impaired osteoblast differentiation. Mechanistically, Padi2 depletion significantly reduces RUNX2 levels, as PADI2-dependent stabilization of RUNX2 protected it from ubiquitin-proteasomal degradation. Furthermore, we discovered that PADI2 binds to RUNX2 and citrullinates it, and identified ten PADI2-induced citrullination sites on RUNX2 through high-resolution LC-MS/MS analysis. Among these ten citrullination sites, the R381 mutation in mouse RUNX2 isoform 1 considerably reduces RUNX2 levels, underscoring the critical role of citrullination at this residue in maintaining RUNX2 protein stability. In conclusion, these results indicate that PADI2 plays a distinct role in bone formation and osteoblast differentiation by safeguarding RUNX2 against proteasomal degradation. In addition, we demonstrate that the loss-of-function of PADI2 is associated with CCD, thereby providing a new target for the treatment of bone diseases.
Similar content being viewed by others


Introduction
Peptidylarginine deiminases (PADIs) are calcium-dependent hydrolases that convert peptidyl-arginine to peptidyl-citrulline, a process known as protein citrullination or deimination. This post-translational modification (PTM) can change the functions of the modified proteins, in terms of protein-protein interactions, protein stability, and subcellular localization, owing to the change from positive to neutral charges [1,2,3,4]. Citrullination regulates several important physiological processes such as early embryogenesis [5], pluripotency of stem cells [6], oligodendrocyte differentiation [7], osteoblast differentiation, and senescence [8]. Abnormal citrullination is closely associated with human diseases, including rheumatoid arthritis, multiple sclerosis, and cancer [9,10,11]. Five PADI isozymes (PADI1, 2, 3, 4, and 6) have been identified in mammals, which have different tissue distributions and overlap** substrates [12]. Our recent study showed that PADI2 is the most prevalent PADI isozyme in osteoblasts and mesenchymal stromal cells and that its downregulation following oxidative stress or Padi2 knockdown inhibits osteoblast differentiation and induces cellular senescence [8]. However, the in vivo function of PADI2 in skeletal formation has not been reported.
Runt-related transcription factor 2 (RUNX2) is an essential master transcription factor in skeletogenesis [13]. Runx2 knockout mice or C-terminal deletion mice exhibit a complete absence of mineralized bone in the calvaria and long bones, suggesting that RUNX2 is required for intramembranous and endochondral bone formation [13,Criteria for protein identification Scaffold (version Scaffold_4.11.0, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability. Peptide Probabilities from X! Tandem were assigned by the Scaffold Local FDR algorithm. Peptide Probabilities from Sequest were assigned by the Peptide Prophet algorithm [27] with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 95.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [28]. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. For statistical analyses, P values were calculated by unpaired two-tailed Student’s t-test (when comparing only two groups), one-way ANOVA, or two-way ANOVA (when comparing more than two groups) using GraphPad Prism 9. All results are expressed as the mean ± SD, and differences were considered significant at P < 0.05. P values are as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. To ensure data reliability, all experiments were performed as at least two or three independent experiments with three replicates. Representative results are shown in the figures.Statistical analysis
Results
Padi2-deficient mice displayed decreased bone mass
Our recent study showed that PADI2 expression levels increased with osteoblast differentiation which was significantly inhibited by Padi2 knockdown, indicating that PADI2 plays an important role in osteoblast differentiation [8]. To understand the role of PADI2 in the bone, we generated global Padi2 knockout mice by crossing Padi2fl/fl with EIIA-Cre mice, a transgenic line in which Cre-mediated recombination occurs in a wide range of tissues, including germ cells that transmit genetic alterations to the progeny [29]. Since EIIA-Cre mice had no significant effect on phenotypes including body size and bone structure compared to wild type mice based on accumulated in-house data and were not significantly different from Padi2fl/fl and Padi2fl/+ mice in bone phenotype, all of EIIA-Cre;Padi2+/+, Padi2fl/fl and Padi2fl/+ mice were used as littermate controls referred to as ‘control’. EIIA-Cre;Padi2fl/+ and EIIA-Cre;Padi2fl/fl mice were named Het and KO mice, respectively. Padi2 KO mice did not show severe phenotypic abnormalities compared to control mice at birth and survived normally. However, the physical size of Padi2 KO mice tended to be smaller than that of their control littermates at the newborn stage and postnatal day seven (P7) (Fig. 1A), and this trend was observed up to 4–5 weeks of age, but there was no significant difference in size between the two groups as they aged further. Body weights of the control and KO groups were consistently similar after 6 weeks of age without significant differences (Supplementary Fig. 1A, B). In male mice, from 14 weeks of age, Padi2 KO mice exhibited a slight but significant weight gain compared to the control (Supplementary Fig. 1A).

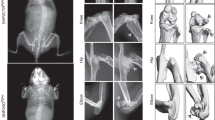
A Physiognomy of Padi2 control (Cont), hetero (Het), and knockout (KO) litters at newborn and P7 (n = 4–6 in each group). B Representative micro-CT images of the distal femur at 4 months in the arterial view of a coronal section and cross-sectional views are shown for each genotype (scale bar:1 mm). C Histomorphometric analyses of 3D micro-CT data of control and Padi2 KO mice in both male (n = 9, Cont mice and n = 7, Padi2 KO mice) and female (n = 11, Cont mice and n = 12, Padi2 KO mice). BV/TV bone volume/tissue volume; Tb. Th trabecular thickness; Tb. N trabecular number; Tb. Sp trabecular separation. D, E Representative images of H&E stained distal femur and proximal tibiae from 4 months-aged control and Padi2 KO mice (scale bar: 500 μm, 200μm). F, G Representative images of immunohistochemistry (IHC) for PADI2 (F) and type 1 collagen (COL1) at 4 months-aged Cont and KO mice (G) (scale bar: 500 μm, 200 μm). H Representative images of TRAP-stained trabecular bone of distal femur from 4 months-aged Cont and KO mice. TRAP-positive purple spots indicate multinucleated osteoclasts (scale bar: 500 μm, 200 μm); We performed two or three independent experiments with three biological replicates for each group (D–H). I Osteoclast identification by TRAP staining. Bone marrow-derived macrophages (BMMs) isolated from Cont and KO mice were differentiated into osteoclasts in the presence of CSF1 (20 ng/ml) and RANKL (100 ng/ml) for 5 days (scale bar: 10 μm). The graph shows the number of TRAP-positive osteoclasts with more than three nuclei (n = 8 in each group). Three independent experiments with eight biological replicates for each group. Data are expressed as the mean ± SE. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. N.S, not significant.
Whole-body Alizarin red/Alcian blue staining revealed no significant defects in the skeletal structures of newborn Padi2 KO mice and respective controls, including the ribs, vertebrae, and limbs. However, delayed mineralization of the calvarial bones and hypoplasia of the clavicles were observed (Supplementary Fig. 2; Fig. 3A). Whole-body micro-computed tomography (μ-CT) analysis showed reduced cranial ossification and a significant decrease in femur length in P7-aged Padi2 KO mice compared to their control littermates (Supplementary Fig. 3A, B). Tibial length exhibited a decreasing tendency in Padi2 KO mice, although no statistically significant decrease was observed compared to the control group (Supplementary Fig. 3B, lower panel). Although PADI2 is the predominant isozyme among the five PADIs in mouse primary calvarial osteoblasts (pOBs) (Supplementary Fig. 4A), other isozymes may compensate for the effect of Padi2 deficiency on skeletal bones. To determine whether other PADI isozymes compensate for the PADI2 deficiency, we compared the mRNA levels of PADI isozymes in pOBs isolated from Padi2 control and KO mice. Interestingly, Padi2 deficiency reduced the mRNA levels of other Padi isozymes (Supplementary Fig. 4B), indicating that the bone phenotypes seen in Padi2 KO mice were not offset by the compensation from other PADI isozymes. To investigate the in vivo effects of PADI2 on the skeletal system of adult mice, we compared the changes in bone-related elements of the distal femur between 4-month-old Padi2 control and KO mice using μ-CT. The Padi2 KO mice showed significantly decreased trabecular bone mass compared to control in males and females (Fig. 1B, C). Trabecular bone per tissue volume (BV/TV) in Padi2 KO mice was lower than that in control, which was accompanied by a reduction in trabecular number and thickness (Fig. 1C). A significant decrease in vertebral bone volume was also observed in 4-month-old Padi2 KO mice, with fewer trabeculae and increased trabecular separation (Supplementary Fig. 5A, B). Long bone sections from 4-month-old Padi2 control and KO mice were subjected to histological analyses. Hematoxylin and eosin (H&E) staining showed that trabecular bones in the femur and tibiae were largely reduced in Padi2 KO mice compared to those in control (Fig. 1D, E). Immunohistochemistry (IHC) showed that while PADI2 was highly abundant in the osteoblasts of control mice, it was almost absent in the femur of Padi2 KO mice (Fig. 1F). In addition, the level of type 1 collagen α1 (COL1A1), a representative osteoblast marker, was significantly lower in Padi2 KO mice than in the control (Fig. 1G).
Tartrate acid resistant phosphatase (TRAP) staining was performed to assess the effects of Padi2 knockout on osteoclast. TRAP-positive multinucleated cells were significantly increased on the surface of the trabecular bone in P7- and 4-month-old Padi2 KO mice compared to that in the control (Supplementary Fig. 6A; Fig. 1H). To investigate whether the increased osteoclastogenesis in Padi2 KO mice was due to Padi2 deficiency, bone marrow cells were collected from the femoral bones of 2-month-old Padi2 control and KO mice and differentiated into osteoclasts. RT-qPCR data demonstrated that early and late osteoclast differentiation markers, including Pu1, Nfatc1, Ctsk, Trap, and Mmp9 significantly increased in Padi2 KO osteoclasts compared to those in control (Supplementary Fig. 6B). TRAP staining revealed that osteoclastogenesis in Padi2 KO bone marrow macrophages (BMMs) was significantly increased compared to that from control BMMs (Fig. 1I). Based on these findings, Padi2 deficiency results in significantly reduced bone mass due to impaired bone formation and increased osteoclastogenesis.
Osteoblastic Padi2 deficiency caused the reduced bone mass with impaired osteoblast differentiation
To further determine the role of PADI2 in bone formation, we generated osteoblast-specific Padi2 knockout mice (hereafter referred to as Padi2Col1 mice) by crossing Padi2fl/fl and Col1α1(2.3 kb)-Cre mice, and Col1α1(2.3 kb)-Cre;Padi2+/+, Padi2fl/fl and Padi2fl/+ mice were all referred to as control. The body size of 4-week-old Padi2Col1 mice was smaller than that of the control (Fig. 2A). The μ-CT scan showed that 4-week-old Padi2Col1 mice displayed a significant decrease in bone mass compared to control (Fig. 2B). Further analysis indicated that BV/TV was significantly decreased in Padi2Col1 mice relative to the control, accompanied by a reduction in trabecular number and an increase in trabecular separation (Fig. 2C). Immunostaining of the distal femur confirmed that PADI2 was completely deficient in the osteoblasts of Padi2Col1 mice, while PADI2 expression was high in the osteoblasts of control mice (Fig. 2D). PADI2 was detected in the bone marrow cells of both the control and Padi2Col1 mice (Fig. 2D). These findings provide evidence of osteoblast-specific Padi2 deficiency in Padi2Col1 mice. Consistent with the μ-CT data, H&E staining also showed diminished trabecular bone loss in the distal femur of Padi2Col1 mice compared to that in the control (Fig. 2E). Consistently, whereas Padi2 and representative bone marker genes, including Runx2, Alp, Bsp, and Ocn showed an increase in pOB from the control group, in a differentiation stage-dependent manner, their mRNA levels were significantly decreased in the pOBs from Padi2 KO mice (Fig. 2F). In addition, the osteoblast differentiation capability of pOBs isolated from the Padi2 control, Het, and KO mice, correlated well with the Padi2 gene dosage determined by alkaline phosphatase (ALP) and alizarin res S (ARS) staining (Fig. 2G). The lower the Padi2 expression level, the greater the inhibition of osteoblast differentiation. Interestingly, Padi2Col1 mice also showed significantly increased numbers of TRAP-positive osteoclasts relative to control mice (Fig. 2H), indicating that secretory factors from Padi2-deficient osteoblasts may stimulate osteoclastogenesis. To determine whether Padi2 deficiency induces osteoclastogenic factors in pOB, we examined the mRNA levels of Csf-1, Rankl, and Opg using RT-qPCR. Csf-1 mRNA levels were significantly increased in pOBs from Padi2 KO mice compared to those from the control (Supplementary Fig. 7A). An increased Rankl/Opg ratio was observed in the pOB from Padi2 KO relative to that from the control owing to increased Rankl mRNA levels (Supplementary Fig. 7A). Our previous study showed that Padi2 knockdown in MC3T3-E1 osteoblasts induced increased mRNA expression and secretion of CCL2, CCL5, and CCL7, which are known to promote osteoclastogenesis [8]. Similarly, the mRNA levels of Ccl2, Ccl5, and Ccl7 were significantly increased in pOBs from Padi2 KO compared to those from control (Supplementary Fig. 7B). Taken together, these results demonstrate that PADI2 is required for bone formation and osteoblast differentiation and that Padi2 deficiency in osteoblasts can promote osteoclastogenesis.

A Representative view of 4-week-old female Cont and Padi2-KOCol1 mice. B Representative micro-CT images of the distal femur from 4-week-old female Cont and Padi2Col1 mice in midsagittal and coronal views. Scale bar: 500 μm. C Histomorphometric analysis of 3D micro-CT of the distal femur from data 4-week-old female Cont (n = 10) and Padi2Col1 (n = 6) mice. D IHC for PADI2 of distal femur from 4-week-old Cont and Padi2Col1 mice. Three independent experiments with three biological replicates for each group. Scale bar: 200 μm, 100 μm. E Representative images of H&E staining for distal femur from 4-week-old Cont and Padi2Col1 mice. Three independent experiments with three biological replicates for each group. Scale bar: 500 μm, 200 μm. F Relative mRNA expression of Padi2 and bone marker genes in Cont and KO primary calvaria osteoblasts (pOBs) cultured or not (Day0) in osteogenic medium for each indicated day as determined by RT-qPCR. Three independent experiments with three biological replicates for each group. G ALP and ARS staining were performed in Cont, Het, and KO primary calvaria OBs cultivated in an osteogenic medium for 5 days and 21 days, respectively. Representative images from three independent experiments with two biological replicates for each group. H TRAP staining of distal femurs from 4-week-old Cont and Padi2Col1 mice. The boxed region is shown in higher magnification (bottom). Three independent experiments with three biological replicates for each group. Scale bar: 500 μm, 200 μm. Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. N.S, not significant. ND not determined.
Ablation of Padi2 causes a CCD-like phenotype
CCD is an autosomal-dominant human bone disease characterized by hypoplastic clavicles, patent fontanelles and sutures, and multiple other skeletal disorders [16]. As mentioned earlier, Alizarin red and Alcian blue staining showed that although newborn Padi2 KO mice did not show significant defects in skeletal structures compared to the control, Padi2 KO mice exhibited a CCD-like phenotype with hypomineralization of the calvarium and clavicular hypoplasia (Fig. 3A). In addition, hypomineralization of calvarial bones was confirmed in both global Padi2 KO mice and osteoblast-specific Padi2 deleted Padi2Col1 mice at P7 (Fig. 3B, C). The μ-CT scanning also confirmed that the clavicle lengths of Padi2 KO mice, which were measured between the sternal end (a) and the conoid tubercle (a’) of the body of the clavicle, were significantly shorter than those of the control (Fig. 3D). CCD is genetically linked to a mutation in RUNX2 [30] and Runx2 haploinsufficiency is known to cause CCD. IHC demonstrated that compared to the control group, global Padi2 KO mice and Padi2Col1 mice showed dramatically reduced RUNX2 levels in the trabecular and cortical bones of the distal femur (Fig. 3E, F). Primary osteoblasts from Padi2 control, Het, and KO mice cultured in osteogenic media exhibited a substantial decrease in RUNX2 protein levels depending on the amount of PADI2 (Fig. 3G). Additionally, Padi2 knockdown using Padi2-specific siRNA induced the significantly reduced RUNX2 levels (Fig. 3H). This phenomenon was also confirmed in the CRISPR–Cas9-mediated Padi2 KO cell clones (#3-4 and #5-6) and WT control cells (Fig. 3I). In contrast, forced expression of Padi2 significantly increased RUNX2 levels in MC3T3-E1 cells cultured in osteogenic medium (Fig. 3J). Collectively, these results suggest that Padi2 deficiency in osteoblasts reduces RUNX2 protein levels, resulting in a CCD-like phenotype.

A Whole-mount skeleton staining of Cont (n = 6) and Padi2 KO (n = 5) newborn littermates by Alizarin red and Alcian blue staining. Samples were cut into calvaria and clavicles. Scale bar: 2 mm. B Representative micro-CT images of the skull of postnatal day 7 (P7) Cont (n = 9) and Padi2 KO (n = 9) mice. Scale bar: 2 mm. C Representative micro-CT images of the skull of P7-old Cont (n = 6) and Padi2 Col1 (n = 5) mice. Scale bar: 2 mm. D The landmarks of the mouse clavicle for measuring the length of the clavicle (left). The clavicle length (a-a’) was measured and graphed in each sample in P7 Cont (n = 8) and Padi2 KO mice (n = 9) (right). E IHC for RUNX2 of the distal femur from 4-week-old Cont and Padi2 KO mice. The second and third rows show enlarged areas of metaphysis and cortical bone, respectively. Three independent experiments with three biological replicates for each group. Scale bar: 500 μm, 200 μm. F IHC for RUNX2 of distal femur from 4-week-old WT and Padi2Col1 mice. The bottom shows cortical bone. Three independent experiments with three biological replicates for each group. Scale bar: 200 μm. G Primary calvarial OBs were cultured in an osteogenic medium for the indicated day, and RUNX2 and PADI2 levels were examined by western blot analysis. α-Tubulin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with α-Tubulin. H MC3T3-E1 cells were transfected with scrambled control siRNA (siCont), Padi2 siRNA #2 (siPadi2 #2), or Padi2 siRNA #3 (siPadi2 #3) and then cultivated in osteogenic media for additional 3 days. RUNX2 and PADI2 levels were examined by western blot analysis. α-Tubulin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with α-Tubulin. I RUNX2 and PADI2 levels in CRISPR–Cas9-mediated Padi2 KO cell clones (#3–4 and #5–6) and control cells were examined by western blot analysis. α-Tubulin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with α-Tubulin. J MC3T3-E1 cells were transfected with empty vector or Flag-PADI2 plasmids and then cultivated in osteogenic media for an additional 3 days. RUNX2 and Flag-PADI2 levels were examined by western blot analysis. GAPDH was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with GAPDH. Western blot data was collected from at least two or three independent experiments; the representative results are shown here.
PADI2 stabilized RUNX2 from ubiquitin-proteasomal degradation
PADI2 is a post-translational modifying enzyme that converts peptidyl-arginine residues to citrulline via deimination, resulting in profound changes in the structure and function of target proteins including protein stability [2]. Padi2 deficiency significantly decreased both RUNX2 mRNA and protein levels (Fig. 2F; Fig. 3E–I). To investigate whether PADI2 affected Runx2 mRNA levels at the post-transcriptional level, MC3T3-E1 cells transfected with Strep-Runx2 with or without Flag-PADI2 were treated with Actinomycin D (ActD), a transcription inhibitor, during the indicated time points. RT-qPCR showed a similar decrease in Runx2 mRNA levels after ActD treatment over time in both the control and PADI2 overexpression groups (Fig. 4A). However, the RUNX2 protein levels significantly decreased 3 h after ActD treatment in the group transfected with the empty vector, whereas it remained the same until 3 h after ActD treatment in the PADI2 overexpressing group (Fig. 4B, C). These results indicated that PADI2 is involved in the regulation of RUNX2 protein levels rather than Runx2 mRNA stability. Next, we investigated whether PADI2 was involved in regulating the stability of RUNX2 by treatment with cycloheximide (CHX), a protein synthesis inhibitor. The RUNX2 protein level was maintained for up to 6 h after CHX treatment when Padi2 was overexpressed. However, the RUNX2 level in the control group showed a time-dependent decrease after CHX treatment (Fig. 4D, E), indicating that PADI2 enhances the half-life of RUNX2 at the post-translational level. Next, we examined whether the Padi2 depletion-induced decrease in RUNX2 was mediated by the ubiquitin-proteasome pathway. Treatment with MG132, a proteasome inhibitor, restored the reduced level of RUNX2 in Padi2 KO pOB cells (Fig. 4F) as well as in cells with Padi2 knockdown or CRISPR-Cas9-mediated Padi2 knockout (Fig. 4G; Supplementary Fig. 8). This restorative effect was also observed in human mesenchymal stromal cells (hMSCs) (Fig. 4H), indicating that the role of PADI2 in regulating the stability of RUNX2 is consistent in human mesenchymal-derived cells. An ubiquitin-based immuno-precipitation assay confirmed that PADI2 overexpression drastically blocked the ubiquitination level of RUNX2 (Fig. 4I). Taken together, these results demonstrate that PADI2 contributes to the maintenance of RUNX2 stability by inhibiting ubiquitination-mediated proteasomal degradation of RUNX2.

A–C MC3T3-E1 cells were transfected with Strep-Runx2 with or without Flag-PADI2, and then cultivated in osteogenic medium for 3 days. On day 3, 4 μg/mL Actinomycin D was treated and incubated for 0, 3, and 6 h. The half-life of Runx2 mRNA and RUNX2 protein was determined by RT-qPCR (A) and western blot analysis (B), respectively. The intensities of Strep-RUNX2 protein levels were normalized against each GAPDH by ImageJ. The normalized values at 0 h were set as 1, and relative levels are shown (C). D, E MC3T3-E1 cells were transfected with Strep-Runx2 together with or without Flag-PADI2, and then cultivated in osteogenic medium for 3 days. 20 μg/mL cycloheximide was treated on the last day and incubated for 0, 3, and 6 h. The half-life of RUNX2 protein was determined by western blot analysis (D). The intensities of Strep-RUNX2 protein levels were normalized against each GAPDH by ImageJ and relative levels are shown (E). F Padi2 control and KO pOB cells were cultured in osteogenic media for 2 days, and 20 μM MG132 or DMSO as vehicle was treated for 6 h before harvesting cells, and western blot analysis followed. β-Actin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with β-Actin. The red arrow indicates PADI2. G MC3T3-E1 cells were transfected with siCont or siPadi2 #2 and then cultivated in osteogenic media for an additional 2 days. 20 μM MG132 or DMSO as the vehicle was treated for 6 h before harvesting cells followed by western blot analysis. α-Tubulin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with α-Tubulin. The red arrow indicates PADI2. H hMSCs were transfected with siCont or siPADI2 and then cultivated in osteogenic media for an additional 2 days. 20 μM MG132 or DMSO as the vehicle was treated for 6 h before harvesting cells followed by western blot analysis. β-Actin was used as a loading control. RUNX2 level was quantified using ImageJ software and normalized with β-Actin. The red arrow indicates PADI2. Western blot data were collected from at least two or three independent experiments; the representative results are shown here. I Strep-Runx2, Flag-PADI2, and HA-ubiquitin were transfected into 293 T cells. 3 days after transfection, cells were treated with 20 μM MG132 for 6 h, lysed, immunoprecipitated with Strep-Tag II magnetic beads, and immunoblotted with anti-HA or anti-RUNX2 antibody. Ubiquination assay was performed in three independent experiments; the representative results are shown here.
PADI2 citrullinates RUNX2
Since PADI2 protected RUNX2 from proteasomal degradation, we further investigated whether PADI2 interacted with RUNX2 for the citrullination of the protein. We transfected 293 T cells with HA-Runx2 with or without Flag-PADI2 plasmids and performed a co-immunoprecipitation (Co-IP) assay, which revealed a protein-protein interaction between PADI2 and RUNX2 (Fig. 5A). Interestingly, in the Co-IP experiment, PADI2 overexpression retarded the movement of some HA-tagged RUNX2 proteins (red arrow in Fig. 5A), suggesting citrullination of RUNX2 by PADI2. Next, to investigate whether PADI2 citrullinates RUNX2, recombinant human RUNX2 protein (rhRUNX2, NP_004339) was in vitro citrullinated by recombinant human PADI2 (rhPADI2) and then labeled with biotin-phenylglyoxal (Biotin-PG), a chemical probe that selectively binds to peptidyl-citrulline under acidic conditions [26, 31, 32]. Sequentially, the labeled proteins were subjected to SDS-PAGE and electro-transferred to the membranes. Streptavidin-HRP was used to detect citrullinated RUNX2 and western blot analysis was performed with an anti-RUNX2 antibody to confirm the presence of both citrullinated and non-citrullinated forms of RUNX2 (Fig. 5B, left panel). The in vitro citrullination assay showed that rhRUNX2 was citrullinated by rhPADI2, and the size of the citrullinated RUNX2 (cit-RUNX2, red asterisk) was slightly larger than that of non-citrullinated RUNX2 (Fig. 5B, middle and right panels). We investigated the effect of citrullination of RUNX2 by PADI2 on the function and fate of RUNX2. Initially, we determined the specific site of citrullination of RUNX2 by PADI2. To accomplish this, we generated cit-rhRUNX2 (NP_004339, 507 aa) by performing in vitro citrullination using rhPADI2. Subsequently we conducted in-gel digestion and high-resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses to identify citrullination site (Supplementary Fig. 9A). Citrullinated forms of seven peptides containing R11, R12, R211, R214, R215, R360 and R372 were identified in the lower cit-RUNX2 band (blue arrow in Supplementary Fig. 9A), and three additional sites (R167, R172, and R176) were identified in the upper cit-RUNX2 band along with the above-mentioned seven sites (red arrow in Supplementary Fig. 9A), compared to native RUNX2 (Fig. 5C; Supplementary Fig. 9A, B). Manual interrogation of the high-resolution MS1 spectra confirmed the presence of the about 1 Da heavier citrullinated species for each of these peptides (Supplementary Fig. 9B). To assess the impact of these citrullination sites on the function and fate of RUNX2, we introduced substitutions in which each of the 10 arginine (R) sites citrullinated by PADI2 was replaced with lysine (K). This substitution was chosen because the conversion from R to K maintains a positive charge but prevents citrullination. Since mouse RUNX2 isoform 1 (mRUNX2, NP_001139510, 528 aa) is the osteoblast-specific isoform and its amino acid sequence is highly conserved with human RUNX2 isoform c, which has a high homology of approximately 95% (Supplementary Fig. 10), it was utilized for mutagenesis. In Fig. 5C, each R site in mouse RUNX2 isoform 1 that corresponds to the 10 citrullinated R sites in human RUNX2 isoform c is depicted, and these R sites were converted to K.

A 293 T cells were transfected with 3xHA-Runx2 together with or without Flag-PADI2 and cultured for 3 days after the transfection. Cells were lysed, immunoprecipitated with anti-HA antibody and protein G-conjugated magnetic beads, and immunoblotted with indicated antibodies. The red arrow indicates predicted citrullinated RUNX2. Co-IP experiment was performed in three independent experiments; the representative results are shown here. B Workflow showing the detection of citrullinated RUNX2 by Biotin-PG labeling (left). Recombinant human RUNX2 (rhRUNX2) isoform c (NP_004339) was in vitro citrullinated by rhPADI2. The samples were labeled with Biotin-PG and then separated by SDS-PAGE followed by transfer to PVDF membrane. The membrane was incubated with Streptavidin conjugated with horseradish peroxidase (Streptavidin-HRP) or was immunoblotted with anti-RUNX2 antibody (right). A red asterisk indicates citrullinated RUNX2. This experiment was performed in three independent experiments; the representative results are shown here. C In vitro citrullinated rhRUNX2 isoform c was analyzed by LC-MS/MS. The ten arginine (R) sites of rhRUNX2 citrullinated by PADI2 were identified (left column). The mouse RUNX2 isoform 1 was used for site-directed mutagenesis of the 10 R sites and, for this, the R site in mouse RUNX2 isoform 1 matching the corresponding each R site in hRUNX2 isoform c is shown in the right column.
PADI2-mediated citrullination of RUNX2 is required for the maintenance of RUNX2 stability
RUNX2 contains several functional domains. Citrullinated R25 and R26 of mRUNX2 were located in the activation domain, six sites (R188, R193, R197, R232, R235, and R236) were within the Runt domain, and R381 and R393 were within the PST domain (Supplementary Fig. 11). To investigate which sites play a key role in the maintenance of RUNX2 protein stability, MC3T3-E1 cells transfected with Strep-mRunx2 wild-type (WT) or mutant forms were cultivated in osteogenic media, and RUNX2 levels were analyzed by western blotting. Interestingly, mRUNX2 R381K mutation among the ten R to K mutations dramatically decreased RUNX2 levels compared to RUNX2 WT (Fig. 6A). To further validate the effect of R381K mutation on RUNX2 stability, WT or R381K mutant constructs were introduced into pOB cells and hMSCs. Western blot analysis showed that mutation in the mRUNX2 R381 locus also greatly reduced RUNX2 levels in both cells (Fig. 6B, C). Core binding factor β (Cbfβ) conditional knockout mice have shown that Cbfβ is required for osteoblast differentiation [22, 33, 34]. Previous studies have demonstrated that Cbfβ is important in the stabilization of RUNX2 by protecting it from degradation by ubiquitination [22, 34]. Cbfβ interacts with the Runt domain that is highly conserved in the RUNX family proteins [35]. Because six sites (R188, R193, R197, R232, R235, and R236) citrullinated by PADI2 were located within the Runt domain of mRUNX2 (Fig. 5C; Supplementary Fig. 11), we investigated whether the ten R mutations, including these six R sites, affected the heterodimerization of RUNX2 with Cbfβ. 293 T cells were transfected with Strep-mRunx2 WT or mutant forms with or without Myc-Cbfβ. Since the expression levels of RUNX2 variants were slightly different, co-immunoprecipitation (Co-IP) was performed after adjusting the expression level of Strep-RUNX2 proteins similarly. Co-IP experiments showed that none of the RUNX2 mutations suppressed the dimerization of RUNX2 with Cbfβ (Fig. 6D). RUNX2 R25K, R26K, and R236K appeared to interact more strongly with Cbfβ than the WT, but this was thought to be due to the higher expression levels of Strep-RUNX2 and Myc-Cbfβ in the Co-IP samples of these mutant groups than in the WT control group. Next, we examined whether these mutations affected the nuclear localization of RUNX2, which functions as a transcription factor. Mutations located near the nuclear localization signal (NLS) (R232K, R235K, and R236K) or near the nuclear matrix target signal (NMTS) (R381K and R393K) did not significantly affect the nuclear localization of RUNX2 (Fig. 6E). The other remaining RUNX2 mutations also did not affect the nuclear localization of the RUNX2 protein (Supplementary Fig. 12). Taken together, these results demonstrate that the citrullination of R381 of RUNX2 plays a critical role in maintaining RUNX2 stability. However, these mutations did not significantly affect the binding of RUNX2 to Cbfβ or the nuclear accumulation of RUNX2.

A MC3T3-E1 cells were transfected with empty vector (EV), Strep-Runx2 wild type (Wt), or R-to-K mutants and cultured in osteogenic media for 3 days after transfection. Cells were lysed and western blot analysis was performed. β-Actin was used as a loading control. Strep-RUNX2 level was quantified using ImageJ software and normalized with β-Actin. The arrow indicates non-specific bands (n.s). Western blot data were collected from three independent experiments; the representative results are shown here. B, C pOB cells and hMSCs were transfected with EV, Strep-Runx2 WT, or R381K mutant plasmids and cultured in osteogenic media for 2 days. Cells were lysed and western blot analysis was performed. β-Actin was used as a loading control. Strep-RUNX2 level was quantified and normalized with β-Actin using ImageJ software. Western blot data were collected from two independent experiments; the representative results are shown here. D 293 T cells were transfected with Strep-Runx2 Wt or R-to-K mutants with or without Myc-Cbfβ plasmids and then incubated for 3 days after transfection. Cells were lysed, immunoprecipitated with Strep-Tag II magnetic beads, and immunoblotted with indicated antibodies. Co-IP experiment was performed in three independent experiments; the representative results are shown here. E MC3T3-E1 cells were transfected with Strep-Runx2 Wt or R-to-K mutants and cultured for 2 days after transfection. Cells were fixed with 4% PFA, permeabilized, and then immunofluorescent staining was performed using anti-Strep-Tag II antibody. DAPI was used for the nucleus. Three independent experiments were performed and the representative results are shown here. Scale bar, 20 μm.
Discussion
In the field of bone biology, citrullination has received less attention compared to other post-translational modifications. However, in autoimmune diseases such as rheumatoid arthritis, it is known that overactivation of PADIs can lead to citrullination of specific proteins like fibrinogen and vimentin, triggering an immune response that contributes to the disease [36, 37]. While previous studies have highlighted the association of PADI overexpression or hyperactivity with pathophysiology, little is known about the role of PADIs and protein citrullination in normal physiological bone tissue. In our study, we demonstrated that PADI2 plays a crucial role in osteoblast differentiation and the communication between osteoblasts and osteoclasts, which is vital for maintaining bone homeostasis. We also observed that PADI2 deficiency leads to bone loss and a human CCD-like phenotype. Moreover, we discovered that PADI2-mediated citrullination of RUNX2, an essential transcription factor that regulates osteoblast differentiation and function, contributes to the stabilization of RUNX2.
Here, EIIA-Cre-mediated global Padi2 KO mice exhibited reduced osteoblast differentiation and increased osteoclastogenesis, which resulted in reduced bone mass. Padi2-deficient BMMs are accelerated into multinucleated mature osteoclasts in these global Padi2 KO mice. These results suggest that Padi2 deficiency in osteoblasts reduces osteoblast function, but its deficiency in BMMs promotes differentiation into osteoclasts, resulting in reduced bone formation, accelerated bone destruction, and ultimately increased bone loss. Osteoblast-specific Padi2 deletion using Col1α1(2.3 kb)-Cre transgenic mice also increased TRAP-positive osteoclasts compared to control mice, indicating that soluble factors secreted from Padi2-deficient osteoblasts are also involved in promoting osteoclastogenesis. Our data showed a significant increase in representative osteoclastogenic factors Csf-1 and Rankl mRNA levels in Padi2 KO pOB cells compared to control cells. However, in our data, RUNX2, known as a key transcription factor regulating the expression of these genes [38, 39], was greatly reduced when Padi2 was deficient. Therefore, the increase in Csf-1 and Rankl transcripts in Padi2-deficient pOBs was possibly promoted by other transcription factors activated by Padi2 depletion. NF-κB p65 transcription factor is one of the candidates involved in this regulation. NF-κB p65 is activated by Padi2 knockdown in osteoblasts [8] and NF-κB p65 binds to the M-CSF promoter in myeloid cell lines [40]. Also, NF-κB signaling pathway mediates HGF-promoted RANKL expression in osteoblasts and bone marrow stromal cells [41]. In addition to these genes, NF-κB promotes the expression and secretion of the senescence-associated secretory phenotype (SASP) factors CCL2, CCL5, and CCL7 in Padi2-deficient osteoblasts [8], which can promote the recruitment of monocytes and osteoclastogenesis [42,43,44]. The inhibition of NF-κB signaling pathway using a pharmacological inhibitor or RNAi significantly reduced the upregulated levels of these genes by Padi2 knockdown in osteoblasts [8], suggesting that blocking NF-κB signaling pathway can be a therapeutic target that can reduce the abnormally increased osteoclastogenic factors and restore the function of osteoblasts lost by reduced PADI2.
The PADI family of enzymes consists of five isozymes (PADI1-4 and PADI6), which exhibit unique tissue localization and have overlap** substrate specificities. These isozymes show high homology among both orthologs and paralogs, with amino acid identities ranging from 44% to 58% among human PADI paralogs [45, 46]. Among these isozymes, PADI2 is the predominantly expressed isozyme in mesenchymal cell-derived osteoblasts, and its loss accelerates cellular senescence and severely inhibits osteoblast differentiation, suggesting that PADI2 plays an important role in osteoblast differentiation and function [8]. However, contrary to expectations, severe developmental bone defects were not observed in these mice although Padi2 KO mice displayed a CCD phenotype and bone loss. The mRNA levels of other PADI isozymes, which were suspected to compensate for the loss of PADI2, were actually lower in Padi2 KO pOB cells compared to control cells. This indicates that the bone phenotype observed in Padi2 KO mice was not due to compensation by other isozymes. Furthermore, our previous study showed that the expression of PADI2 was low during osteoblast proliferation but increased as differentiation progresses, and loss of PADI2 due to oxidative stress has been shown to induce DNA damage and the SASP factors, leading to osteoblast senescence [8]. These findings suggest that PADI2 is important for defending against aging-associated oxidative stress and maintaining cellular homeostasis. Therefore, it is believed that PADI2 may play a more significant role in maintaining bone homeostasis in mature or aged bone rather than during early bone development. Collectively, our previous and current studies highlight the importance of PADI2 in osteoblast differentiation, bone homeostasis, and defense against oxidative stress. Thus, its deficiency leads to bone loss and compromises the maintenance of bone integrity, particularly in mature or aged bone.
RUNX2 is regulated by various post-translational modifications. The fibroblast growth factor (FGF)/FGF receptor (FGFR) and bone morphogenetic protein (BMP)/BMP receptor (BMPR) signaling pathways, which are essential for osteoblast proliferation and differentiation, induce the acetylation and phosphorylation of RUNX2, which enhances its stability and transcriptional activity [18, 19]. Furthermore, the acetylation of RUNX2 by FGF2 requires the phosphorylation of RUNX2 by ERK MAPK and the subsequent isomerization of RUNX2 by PIN1, which recognizes the phosphorylation of RUNX2 [47]. However, the role and regulatory mechanism of PADI enzyme-mediated citrullination in osteoblast differentiation and function have not been extensively studied until now. Also, the involvement of citrullination in regulating key factors of osteoblast differentiation, such as RUNX2, has not been reported. This study provides novel insights by demonstrating that PADI2 is involved in protecting RUNX2 from ubiquitin-mediated proteasomal degradation. The findings suggest that the citrullination of RUNX2 by PADI2 plays a crucial role in maintaining the stability of the RUNX2 protein. However, it is important to note that the citrullination of RUNX2 does not act alone but likely cooperates with other PTMs to regulate RUNX2 stability. This is supported by the partial rescue of RUNX2 protein levels observed with MG132 treatment in Padi2 knockout and knockdown cells. Furthermore, citrullination at the R381 site of RUNX2 has been identified as important for maintaining the stability of the RUNX2 protein. This suggests that the citrullination of this particular site is critical for the regulatory function of PADI2 on RUNX2 stability.
Contrary to expectations, site-directed mutagenesis of the citrullination sites identified in this study did not inhibit the heterodimerization of RUNX2 with Cbfβ nor did it affect the nuclear localization of RUNX2. However, although not revealed in this study, citrullination of these R sites in RUNX2 can affect various functions of RUNX2, such as protein-protein interactions, DNA-binding activity, and transcriptional activity. In addition, citrullination of RUNX2 may induce conformational changes in RUNX2, allowing it to better bind to the promoter region of target genes or proteins involved in regulating RUNX2 stability. Further studies are required to answer these questions.
In this study, Padi2 KO mice exhibited a human CCD-like phenotype. CCD is usually caused by haploinsufficiency of RUNX2 due to mutations in humans [48]. Mass spectrometry revealed that PADI2 directly citrullinated 10 R sites within the RUNX2 protein. These results indicate that post-translational citrullination of RUNX2 by PADI2 can modulate RUNX2 function, and its dysregulation can lead to bone diseases, such as CCD. Among the 10 R citrullination sites of RUNX2 identified in this study, missense mutations at sites corresponding to R193, R197, R232, and R381 of mRUNX2 isoform 1 have been reported in human patients with CCD [48,49,50,51,52,53]. In approximately 30 percent of the individuals with CCD, no mutations in the RUNX2 gene have been found. The cause of this condition remains unclear. However, these patients may have a loss-of-functional mutation of the PADI2 gene, which in turn affects RUNX2 function. Here, we show that citrullination of RUNX2 at R381 by PADI2 is an essential post-translational modification for maintaining RUNX2 protein stability. Although a direct correlation between citrullination and the R-site mutations within RUNX2 reported in patients with CCD has not been elucidated, this strongly suggests that citrullination at these R sites may play an important role in RUNX2 function. Taken together, these results suggest that citrullination of RUNX2 is a critical post-translational modification for osteoblast differentiation and function.
Collectively, we highlighted, for the first time, the critical role of PADI2 in bone formation and homeostasis using global and osteoblast-specific conditional Padi2 KO mice, shedding light on its underlying mechanisms. Padi2 deficiency leads to a reduction in bone mass and the development of the CCD phenotype, primarily due to decreased stability of the RUNX2 protein. In vitro mechanistic analyses have demonstrated that PADI2 citrullinates RUNX2 and prevents its proteasomal degradation. This study provides novel evidence elucidating the involvement of PADI2 and its citrullination in osteoblast differentiation and function, thereby opening up new avenues for targeting bone diseases, including CCD and senile osteoporosis, in potential therapeutic interventions
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available from the corresponding author upon request. The authors follow the guidelines provided by the journal for sharing the data. Mass spectrometry proteomics data are available via ProteomeXchange with the identifier PXD040179.
References
Guo Q, Fast W. Citrullination of inhibitor of growth 4 (ING4) by peptidylarginine deminase 4 (PAD4) disrupts the interaction between ING4 and p53. J Biol Chem. 2011;286:17069–78.
Deplus R, Denis H, Putmans P, Calonne E, Fourrez M, Yamamoto K, et al. Citrullination of DNMT3A by PADI4 regulates its stability and controls DNA methylation. Nucleic Acids Res. 2014;42:8285–96.
Sun B, Dwivedi N, Bechtel TJ, Paulsen JL, Muth A, Bawadekar M, et al. Citrullination of NF-kappaB p65 promotes its nuclear localization and TLR-induced expression of IL-1beta and TNFalpha. Sci Immunol. 2017;2:eaal3062.
Abdallah BM, Haack-Sorensen M, Fink T, Kassem M. Inhibition of osteoblast differentiation but not adipocyte differentiation of mesenchymal stem cells by sera obtained from aged females. Bone .2006;39:181–8.
Esposito G, Vitale AM, Leijten FP, Strik AM, Koonen-Reemst AM, Yurttas P, et al. Peptidylarginine deiminase (PAD) 6 is essential for oocyte cytoskeletal sheet formation and female fertility. Mol Cell Endocrinol. 2007;273:25–31.
Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature .2014;507:104–8.
Falcao AM, Meijer M, Scaglione A, Rinwa P, Agirre E, Liang J, et al. PAD2-mediated citrullination contributes to efficient oligodendrocyte differentiation and myelination. Cell Rep. 2019;27:1090–102 e10.
Kim HJ, Kim WJ, Shin HR, Yoon HI, Moon JI, Lee E, et al. ROS-induced PADI2 downregulation accelerates cellular senescence via the stimulation of SASP production and NFkappaB activation. Cell Mol Life Sci. 2022;79:155.
Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81.
Moscarello MA, Mastronardi FG, Wood DD. The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem Res. 2007;32:251–6.
Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer. 2009;9:40.
Darrah E, Rosen A, Giles JT, Andrade F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: novel insights into autoantigen selection in rheumatoid arthritis. Ann Rheum Dis. 2012;71:92–8.
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell .1997;89:755–64.
Choi JY, Pratap J, Javed A, Zaidi SK, **ng L, Balint E, et al. Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci USA. 2001;98:8650–5.
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell .1997;89:765–71.
Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36:177–82.
**ao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, et al. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem. 2000;275:4453–9.
Park OJ, Kim HJ, Woo KM, Baek JH, Ryoo HM. FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J Biol Chem. 2010;285:3568–74.
Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, ** YH, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. 2006;281:16502–11.
Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–66.
Qu Y, Olsen JR, Yuan X, Cheng PF, Levesque MP, Brokstad KA, et al. Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat Chem Biol. 2018;14:94–101.
Lim KE, Park NR, Che X, Han MS, Jeong JH, Kim SY, et al. Core binding factor beta of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. J Bone Min Res. 2015;30:1943.
Park NR, Lim KE, Han MS, Che X, Park CY, Kim JE, et al. Core binding factor beta plays a critical role during chondrocyte differentiation. J Cell Physiol. 2016;231:162–71.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8.
Clancy KW, Weerapana E, Thompson PR. Detection and identification of protein citrullination in complex biological systems. Curr Opin Chem Biol. 2016;30:1–6.
Lewallen DM, Bicker KL, Subramanian V, Clancy KW, Slade DJ, Martell J, et al. Chemical Proteomic Platform To Identify Citrullinated Proteins. ACS Chem Biol. 2015;10:2520–8.
Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92.
Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58.
Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5.
Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997;89:773–9.
Bicker KL, Subramanian V, Chumanevich AA, Hofseth LJ, Thompson PR. Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination. J Am Chem Soc. 2012;134:17015–8.
Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, et al. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem Biol. 2018;25:691–704 e6.
Chen W, Ma J, Zhu G, Jules J, Wu M, McConnell M, et al. Cbfbeta deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfbeta required for skeletal development. Proc Natl Acad Sci USA. 2014;111:8482–7.
Qin X, Jiang Q, Matsuo Y, Kawane T, Komori H, Moriishi T, et al. Cbfb regulates bone development by stabilizing Runx family proteins. J Bone Min Res. 2015;30:706–14.
Wang SW, Speck NA. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol. 1992;12:89–102.
Damgaard D, Senolt L, Nielsen MF, Pruijn GJ, Nielsen CH. Demonstration of extracellular peptidylarginine deiminase (PAD) activity in synovial fluid of patients with rheumatoid arthritis using a novel assay for citrullination of fibrinogen. Arthritis Res Ther. 2014;16:498.
Van Steendam K, Tilleman K, Deforce D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatol (Oxf). 2011;50:830–7.
Yoon H, Kim HJ, Shin HR, Kim BS, Kim WJ, Cho YD, et al. Nicotinamide improves delayed tooth eruption in Runx2(+/−) mice. J Dent Res. 2021;100:423–31.
Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol Cell Biol. 2002;22:6222–33.
Kogan M, Haine V, Ke Y, Wigdahl B, Fischer-Smith T, Rappaport J. Macrophage colony stimulating factor regulation by nuclear factor kappa B: a relevant pathway in human immunodeficiency virus type 1 infected macrophages. DNA Cell Biol. 2012;31:280–9.
Tsubaki M, Seki S, Takeda T, Chihara A, Arai Y, Morii Y, et al. The HGF/Met/NF-kappaB pathway regulates RANKL expression in osteoblasts and bone marrow stromal cells. Int J Mol Sci. 2020;21:7905.
Graves DT, Jiang Y, Valente AJ. The expression of monocyte chemoattractant protein-1 and other chemokines by osteoblasts. Front Biosci. 1999;4:D571–80.
Lee JW, Hoshino A, Inoue K, Saitou T, Uehara S, Kobayashi Y, et al. The HIV co-receptor CCR5 regulates osteoclast function. Nat Commun. 2017;8:2226.
Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Min Res. 2004;19:2065–77.
Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25:1106–18.
Slade DJ, Fang P, Dreyton CJ, Zhang Y, Fuhrmann J, Rempel D, et al. Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design. ACS Chem Biol. 2015;10:1043–53.
Yoon WJ, Cho YD, Kim WJ, Bae HS, Islam R, Woo KM, et al. Prolyl isomerase Pin1-mediated conformational change and subnuclear focal accumulation of Runx2 are crucial for fibroblast growth factor 2 (FGF2)-induced osteoblast differentiation. J Biol Chem. 2014;289:8828–38.
Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19:209–16.
Zhang X, Liu Y, Wang X, Sun X, Zhang C, Zheng S. Analysis of novel RUNX2 mutations in Chinese patients with cleidocranial dysplasia. PLoS One. 2017;12:e0181653.
Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, et al. Functional analysis of RUNX2 mutations in cleidocranial dysplasia: novel insights into genotype-phenotype correlations. Blood Cells Mol Dis. 2003;30:184–93.
Ryoo HM, Kang HY, Lee SK, Lee KE, Kim JW. RUNX2 mutations in cleidocranial dysplasia patients. Oral Dis. 2010;16:55–60.
Lee KE, Seymen F, Ko J, Yildirim M, Tuna EB, Gencay K, et al. RUNX2 mutations in cleidocranial dysplasia. Genet Mol Res. 2013;12:4567–74.
Kim HJ, Nam SH, Kim HJ, Park HS, Ryoo HM, Kim SY, et al. Four novel RUNX2 mutations including a splice donor site result in the cleidocranial dysplasia phenotype. J Cell Physiol. 2006;207:114–22.
Funding
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1A4A1019423 and 2020R1A2B5B02002658 to HMR; 2021R1A2C1007715 and 2021R1A6A1A03039462 to HJK; and 2022R1I1A1A01053914 to HR Shin).
Author information
Authors and Affiliations
Contributions
HJK designed the study, did majority of the experiments, and wrote the manuscript; HRS did majority of the experiments including bone sampling, micro-CT analysis and histological analysis and contributed to writing of the manuscript; HIY performed the isolation of primary OB cells and BMM cells from mice, conducted TRAP staining and RT-qPCR, and contributed to tissue sampling; MSP took care of the mice, did genoty**, measurement of body weight, and RT-qPCR, and contributed to tissue sampling; BKK performed proteomic experiments and analysis and contributed to writing of the manuscript; JIM performed IF assay and contributed to tissue sampling; WJK contributed to the experimental design an contributed to tissue sampling; SGP did statistical analysis; KTK contributed to the experimental design and statistical analysis; HNK provided critical comments for micro-CT and histology data, contributed to the experimental design for animal study and editing the manuscript; JYC provided critical advice for animal study and data analysis and edited the manuscript; HMR supervised the study and wrote the manuscript as the corresponding author. All the authors have read and commented on the manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experiments involving mice were performed with the approval of the Institutional Animal Care and Use Committee and the Special Committee on Animal Welfare, Seoul National University, Seoul, South Korea.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Ying Wang
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, HJ., Shin, HR., Yoon, H. et al. Peptidylarginine deiminase 2 plays a key role in osteogenesis by enhancing RUNX2 stability through citrullination. Cell Death Dis 14, 576 (2023). https://doi.org/10.1038/s41419-023-06101-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06101-7
- Springer Nature Limited