
Abstract
Background
Although PD-1 has been reported to be a marker of T-cell exhaustion in several malignancies, the biological role of PD-1+CD8+ T cells in gastric cancer (GC) remains unclear. Herein, we aimed to investigate the role of PD-1+CD8+ T cells in the tumour microenvironment and its clinical significance in GC.
Designs
This study included 441 tumour microarray specimens and 60 Flow cytometry specimens of GC patients from Zhongshan Hospital, and 250 GC patients from the Asian Cancer Research Group.
Results
Here, we demonstrated that PD-1+CD8+ T cells functioned as an independent adverse prognosticator in GC. In addition, an abundance of intratumoral PD-1+CD8+ T cells indicated worse chemotherapeutic responsiveness to fluorouracil in Stage III GC patients. Mechanistically, PD-1+CD8+ T cell high infiltration indicated an exhausted phenotype of global CD8+ T cells in GC tissues, which was characterised by elevated immune checkpoint expression including CTLA-4 and TIM-3, whereas decreased expression of perforin. Furthermore, PD-1+CD8+ T cell high-infiltration patients with Stage III GC held elevated activity of several therapeutic signal pathways.
Conclusions
Our study highlighted that PD-1+CD8+ T cell abundance predicts inferior prognosis in GC, and may serve as a novel predictive biomarker to guide therapeutic option.
Similar content being viewed by others


Introduction
Gastric cancer (GC) ranks the fifth most frequently diagnosed malignancies and the third major cause of cancer-related mortality worldwide [1,2,3,4]. Generally, radical gastrectomy remains the only curative treatment for GC [5, 6]. Nevertheless, since early-stage GC is asymptomatic, most patients are diagnosed at an advanced stage with dismal clinical outcomes [4]. Adjuvant chemotherapy (ACT) has been routinely applied in advanced-stage GC patients to overcome postoperative recurrence [4], of which 5-fluorouracil (5-FU) has been recognised as one of the standard-of-care agents [7,8,9]. However, restricted survival benefit was achieved due to intrinsic and acquired chemoresistance [10, 11]. Hence, novel GC stratification frameworks to facilitate personalised treatment selection have entered the spotlight.
Tumours are organised ecosystems instead of the simply sum of subclones. Tumour microenvironment (TME) is the “fertile soil” for cancer initiation and progression, which plays a versatile role in determining the biological properties of cancer [12]. Specifically, the immune contexture represents a crucial component of TME, which is shaped by the density, composing, functional status and reciprocal interaction of the tumour-infiltrating immune cells [13]. As we have previously reported, the immune contexture in GC is predictive of both overall prognosis and response to either chemotherapy or immunotherapy [14]. However, while effector immune subsets are responsible for immune scrutiny, tumour cells may also manipulate the host immune system, cause immune evasion and lead to disease progression [15]. As a result, the abundance of several immune subsets, including special subsets of macrophages [16, 17], CD8+ T cells [18] and of CD4+ T cells [19, 20], may even relate to a tumour-promoting TME.
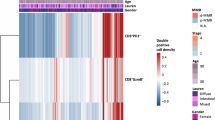
Usually, CD8+ T cells are paramount in the antitumour immune process [21]. Nevertheless, existing studies that examine the prognostic impact of CD8+ T cell infiltration in GC are inconsistent [22], indicating the heterogeneity of intratumoral CD8+ T cells. Contrasting to conventional activated CD8+ T cells that function as the immune effector, persistent antigen stimulation may also lead to a dysfunctional state called T-cell exhaustion [23, 24]. Immune checkpoints are typically overexpressed on exhausted T cells, of which programmed cell death-1 (PD-1) has been recognised as a crucial marker with biological effects to inhibit T-cell activation. Recent studies have revealed that the abundance of PD-1+CD8+ T cells is associated with worse prognosis and impaired antitumour immunity in patients with liver [25], lung [26], ovary [27], colorectum [32, 33]. We found that PD-1+CD8+ T cells were specifically enriched in genomic stable (GS) subtype and EBV-positive subtype according to TCGA classification and epithelial-to-mesenchymal transition (EMT) subtype based on ACRG classification. (Fig. 1b and Supplementary Fig 2A, B). Furthermore, we found that compared with Stage I tumours, Stage III tumours were infiltrated with more PD-1+CD8+ T cells (Fig. 1c; P = 0.042). Together, these findings indicated that PD-1+CD8+ T cells abundance might identify a biologically aggressive subtype of GC.

a Left: representative immunohistochemistry images for tumour-infiltrating PD-1+CD8+ T cells in gastric tissues. PD-1+ cells were stained in brown, while CD8+ cells were stained in red. Cells double-stained in brown and red were recognised as PD-1+CD8+ T cells (black arrows). Magnification: ×200; Scale bar, 100 μm. Right: scatterplots indicated the cumulative frequency of PD-1+CD8+ T cells in the gastric tumour and peritumor tissues (n = 441, Paired t test, P < 0.05). b PD-1+CD8+ T signature score in the EBV, MSI, GS and CIN subgroups (TCGA classification) and in the MSI, MSS/EMT, MSS/TP53− and MSS/TP53+ subgroups (ACRG classification). c Association between PD-1+CD8+ T cells and tumour TNM stage was examined based on IHC staining (P = 0.049, One-way ANOVA followed by Tukey multiple comparisons). CIN Chromosomal instability, EBV EBV-positive, GS genomically stable, MSI microsatellite instable, MSS/EMT microsatellite stable and epithelial-to-mesenchymal transition, MSS/TP53+ microsatellite stable and tumour protein 53 active, MSS/TP53− microsatellite stable and tumour protein 53 inactive. Small horizontal lines indicate the mean (±SD). *P < 0.05, **P < 0.01, ***P < 0.001, ns refers to not significant. ANOVA analysis of variance, PD-1 programmed cell death protein 1, SD standard deviation.
Intratumoral PD-1+CD8+ T-cell infiltration indicated unfavourable prognosis in GC
Consistent with previous studies, we found that the total infiltrating density of intratumoral CD8+ T cells in GC might not be indicative of prognosis (P = 0.135, Supplementary Fig. 1A). This conclusion suggests that relative to overall CD8+ T cell infiltration, specific functional subsets of CD8+ T cells may play a key role in GC. Here, we focused on intratumoral PD-1+CD8+ T cell in GC. Kaplan–Meier curves and log-rank test were conducted to compare the OS and DFS between PD-1+CD8+ T cells high and low infiltration subgroups. In both Discovery Set and Validation Set, abundance of PD-1+CD8+ T cells predicted significantly worse OS (P < 0.001 and P < 0.001; Fig. 2a, b, left panel) and DFS (P = 0.011 and P < 0.001; Fig. 2a, b, right panel). Subsequently, multivariate Cox regression analysis was performed. Clinicopathological parameters, including age, gender, Lauren’s classification, location, tumour grade, tumour size, and TNM stage, along with ACT and PD-1+CD8+ T cells density were incorporated into the multivariate Cox regression model. Consequently, we found that PD-1+CD8+ T cell infiltration predicted poor prognosis independent of the above clinicopathological parameters based on OS (discovery set: hazard ratio (HR) = 2.04, 95% confidence interval (CI) = 1.30–3.19, P = 0.001; validation set: HR = 1.63, 95% CI = 1.05–2.53, P = 0.025; Fig. 2c) and DFS (discovery set: HR = 2.33, 95% CI = 1.57–3.46, P < 0.001; validation set: HR: 2.43, 95% CI: 1.63–3.61, P < 0.001; Fig. 2c). Collectively, these results indicated that the infiltration of PD-1+CD8+ T cells could serve as an independent adverse prognosticator for survival outcomes in GC.

a, b Kaplan–Meier curve of OS (left panel) and DFS (right panel) in discovery set (a) and validation set (b) according to PD-1+CD8+ T cells infiltration. Data were analysed by log-rank test. c Multivariate Cox analysis of OS and DFS for PD-1+CD8+ T cells infiltration and clinicopathologic factors in discovery set and validation set. HR hazard ratio, CI confidence interval, OS overall survival, DFS disease-free survival.
Abundance of intratumoral PD-1+CD8+ T cells predicted inferior efficacy of ACT in TNM Stage III GC
In the ZSHS Cohort, patients with TNM II and III GC could benefit from fluorouracil-based ACT (P < 0.001, HR = 0.49; Fig. 3a). However, the predictive value of intratumoral PD-1+CD8+ T cells regarding ACT remains unclear. Thus, we aimed to explore the efficacy of fluorouracil-based ACT in different subgroups based on PD-1+CD8+ T cells infiltration with univariate Cox regression model. In patients with Stage II/III GC, superior ACT reactivity was shown in both PD-1+CD8+ T cells high subgroup (P = 0.007, HR = 0.57; Fig. 3a) and PD-1+CD8+ T cells low subgroup (P < 0.001, HR = 0.33; Fig. 3a). Therefore, in order to investigate the relationship between PD-1+CD8+ T cell infiltration and benefit from ACT, we further conducted an interaction analysis among subgroups, in which, a significant interaction test shows that the treatment effect significantly varies across the levels of the subgroup. Although no significant results were observed according to interaction test between PD-1+CD8+ T cells infiltration and ACT, in the PD-1+ CD8+ T cells high-infiltration subgroup, we observed a trend of poor response to chemotherapy (P = 0.065 for interaction; Fig. 3a). Consequently, we further stratified patients according to TNM stage, patients were divided into Stage II group (P = 0.020, HR = 0.45; Fig. 3b) and Stage III group (P < 0.001, HR = 0.32; Fig. 3c). In patients with Stage II GC, we found that ACT successfully prolonged OS in patients with PD-1+CD8+ T cells high infiltration (P = 0.019, HR = 0.36; Fig. 3b), while no survival benefit was observed in patients with low PD-1+CD8+ T cells infiltration (P = 0.34, HR = 0.59; Fig. 3b). However, the interaction test suggested that there was no significant difference in ACT effectiveness between the two groups of patients with Stage II GC (P = 0.45 for interaction; Fig. 3b). Therefore, we assumed that the negative results of PD-1+CD8+ T cells low infiltration patients with Stage II GC might result from the relatively small cohort (n = 50). Subsequently, in patients with Stage III GC, ACT provided a significant survival benefit in both PD-1+CD8+ T cells high-infiltration subgroup (P < 0.001, HR = 0.35; Fig. 3c) and PD-1+CD8+ T cells low-infiltration subgroup (P < 0.001, HR = 0.21; Fig. 3c). Interestingly, the interaction test revealed that PD-1+CD8+ T cells high-infiltration patients had worse therapeutic responsiveness to ACT than PD-1+CD8+ T cells low-infiltration patients (P = 0.037 for interaction; Fig. 3c). Herein, these results suggested that PD-1+CD8+ T cells low abundance predicted optimal fluorouracil-based chemotherapeutic responsiveness for patients with TNM Stage III GC, and patients with PD-1+CD8+ T cells high abundance might have a higher risk of chemoresistance.

a Kaplan–Meier curves of OS in patients with TNM Stage II and III GC (n = 382, P < 0.001, HR = 0.49) (left panel), patients with high PD-1+CD8+ T cell infiltration (n = 172, P = 0.007, HR = 0.57) (middle panel) and patients with low PD-1+CD8+ T cells (n = 156, P < 0.001, HR = 0.33) (right panel) according to ACT application. Log-rank test was applied to Kaplan–Meier curves. b Kaplan–Meier curves of OS in patients with TNM Stage II GC (n = 101, P = 0.020, HR = 0.45) (left panel), patients with high PD-1+CD8+ T cell infiltration (n = 51, P = 0.019, HR = 0.36) (middle panel) and patients with high PD-1+CD8+ T cells (n = 50, P = 0.34, HR = 0.59) (right panel) according to ACT application. Log-rank test was applied to Kaplan–Meier curves. c Kaplan–Meier curves of OS in patients with TNM Stage III tumours (n = 227, P < 0.001, HR = 0.32) (left panel), patients with high PD-1+CD8+ T cell infiltration (n = 121, P < 0.001, HR = 0.36) (middle panel) and patients with high PD-1+CD8+ T cells (n = 106, P < 0.001, HR = 0.21) (right panel) according to ACT application. Log-rank test was applied to Kaplan–Meier curves.
Intratumoral PD-1+CD8+ T cells abundance indicated impaired CD8+ T-cell effector function
Since we have highlighted the clinical significance of PD-1+CD8+ T cells, and found PD-1+CD8+ T cells could predict poor OS and inferior chemotherapeutic responsiveness in GC (Figs. 2 and 3), we wondered whether this subgroup of CD8+ T cells was associated with CD8+ T cell dysfunctional phenotype. Thus, FCM analysis was performed on sixty GC patients to detect the proportion of PD-1+CD8+ T cells among CD8+ T cells. Based on the above data, the sixty patients were divided into two subgroups (PD-1+CD8+ T cells high-infiltration and PD-1+CD8+ T cells low infiltration). The global characterisation of CD8+ T cells was subsequently investigated according to PD-1+CD8+ T cell abundance (Fig. 4a). Notably, we found that CD8+ T cells in tumours with PD-1+CD8+ T cells high infiltration expressed increased immune checkpoints, including cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and T-cell immunoglobulin domain and mucin domain-3 (TIM-3) while decreased effector cytokines, perforin 1 (PRF1) (Fig. 4b) than their counterparts. While no significant difference was observed among the two groups, regarding the proliferative ability (Ki-67), the effector cytokines, interferon-γ (INF-γ) and tumour necrosis factor-α (TNF-α) and cytotoxicity activation molecules, Granzyme B (GZMB) expressed by CD8+ T cells (Supplementary Fig. 3B). These findings indicated that terminally exhausted CD8+ T cells were most predominant in PD-1+CD8+ T cells high-infiltration Stage III GC, and high expression of PD-1 on CD8+ T cells were associated with counteracted and impaired CD8+ T cell antitumor immunity. The results aforementioned preliminarily verified our conjecture that intratumoral PD-1+CD8+ T cell abundance might contribute to immune suppression and dampen CD8+ T cell immune response in GC.

a Heatmap illustrating the infiltration of CD8+ T cells with Immune-related molecules high expression by flow cytometry analysis among PD-1+CD8+ T cell infiltration and clinical stage from patients with GC (normalised by the Z-score). b The quantification of exhausted markers (CTLA-4 and TIM-3) and effector cytokines (PRF1) expression on CD8+ T cells in PD-1+CD8+ T cells high/low abundance subgroups, comparisons were also demonstrated among different TNM stages. Data were analysed by Mann–Whitney U test. Small horizontal lines indicate the mean (± SD). *P < 0.05, **P < 0.01, ***P < 0.001, ns refers to not significant. All P values presented here were two-tailed. IFN-γ interferon-γ, GZMB granzyme B, PD-1 programmed cell death protein 1, CTLA-4 cytotoxic T-lymphocyte-associated protein-4, TIM-3 T cell immunoglobulin, LAG3 lymphocyte-activation gene 3.
Abundance of PD-1+CD8+ T cells correlated with actionable genomic alterations
Previous results we acquired had shown that Stage III GC patients with high PD-1+CD8+ T cell infiltration have a higher risk of chemoresistance. In order to improve the survival benefit of these patients, more appropriate treatment strategies need to be considered and may be used as an alternative after chemoresistance.
We sought to inspect the association between PD-1+CD8+ T cell signature and potentially targetable genomic alterations in Stage III GC. Notably, PD-1+CD8+ T cells high subgroup exhibited significant gene enrichment of TGFB signalling, ERBB signalling (human epidermal growth factor receptor) signalling, VEGF/VEGFR (vascular endothelial growth factor) signalling (Fig. 5). The above findings may provide new therapeutic ideas for Stage III GC patients with high PD-1+CD8+ T cell infiltration who are resistant to chemotherapy. Meanwhile, We also found that PD-1+CD8+ T cells low subgroup exhibited significant gene enrichment MUC17 mRNA and HRR signalling. Taken together, our results suggest advanced patients with GC could be divided into two subgroups, which might be sensitive to different therapeutic strategies.

a Heatmap demonstrated the genomic alterations of potential therapeutic targets in GC based on PD-1+CD8+ T cell signature level. b Quantification analyses of therapy-associated signal pathway signature between PD-1+CD8+ T cell signature low/high-expression subgroup among Stage III GC patients in ACRG cohort: TGFB signalling pathway (P < 0.001), VEGF/VEGFR signalling network (P < 0.001), ERBB signalling pathway (P < 0.05), MUC17 relative mRNA expression (P < 0.001), HRR signalling pathway (P < 0.010); HRR, homologous recombination repair. Data were analysed by Mann–Whitney U test. Small horizontal lines indicate the mean (±SD). *P < 0.05, **P < 0.01, ***P < 0.001, ns refers to not significant. P values presented here were two-tailed.
Discussion
Previous results that examine the clinical significance of CD8+ T cells in GC are inconsistent, reflecting the functional complexity of this heterogenous cell population [34]. In this study, we delineated a functional distinct subset of PD-1 expressing CD8+ T cells in GC, and linked PD-1+CD8+ T abundance with immune-evasive TME and unfavourable clinical outcomes.
First, our study again highlighted the heterogeneity of CD8+ T cells in GC, and profiled the phenotypic and functional properties of PD-1 expressing CD8+ T subset. We found that PD-1+CD8+ T cells displayed a dysfunctional phenotype, featured by overexpression of immune checkpoints, including CTLA-4 and TIM-3, yet loss of perforin expression. Specifically, PD-1+CD8+ T cells were especially abundant in EMT/MSS subtype GC according to ACRG classification. The EMT/MSS subtype GC was reported to correlate with low TMB, worst prognosis and high risk of recurrence [32], which may partly account for why PD-1+CD8+ T cell high-infiltration tumours were extremely lethal. As a regulatory pathway to shape the EMT subtype, we also observed elevated TGF-beta signalling in PD-1+CD8+ T abundant tumours, which was found to correlate with worse clinical outcomes and immune-evasive TME in our previous work [14]. Consequently, dual inhibition of PD-1/PD-L1 axis and TGF-beta signalling may be especially rewarding in patients with PD-1+CD8+ T cells high infiltration.
Second, we provided PD-1+CD8+ T as a novel biomarker to select GC patients for both fluorouracil-based ACT and targeted agents. In this study, we found that TNM Stage III GC patients with PD-1+CD8+ T abundance could restrictedly benefit from fluorouracil-based ACT. Thus, PD-1+CD8+ T cells could be a predictive biomarker to select TNM Stage III GC patients for postoperative chemotherapy. Moreover, drugs targeting actionable genomic alterations [35], including HER2 inhibitors and anti-angiogenesis therapies, have been brought into clinical trials of advanced-stage GC [36, 37]. Our results revealed that tumours with PD-1+CD8+ T abundance displayed elevated VEGF/VEGFR and ERBB signalling pathway activity. Therefore, patients with this aggressive subtype GC might potentially benefit from Anti-angiogenesis and HER2 targeted therapies.
Although a previous study by Shen et al. showed that PD-1+CD8+ T cells showed equivalent function to their PD-1-CD8+ T cells counterparts and they did not predict tumour progression in GC, which seemed contradictory with our findings [30]. Actually, PD-1+CD8+ T cells might be also a heterogenous cell subset with functional diversity. Kim HD et al reported that the tumour-infiltrating CD8+ T cells could be subdivided into PD-1-high, PD-1-intermediate, and PD-1-negative subpopulations with distinct gene expression profiles, different exhaustion-related immunophenotypes, and functional capacities [25]. Thus, considering the difference in methods to identify PD-1+CD8+ T cells, it is possible that the main subsets of PD-1+CD8+ T cells might be different in Shen et al’s and our study which might account for the inconsistent findings. And furthermore in-depth research about the exact roles for subsets of PD-1+CD8+ T cells should be conducted in the future.
Several limitations were presented in our current study. First, further studies were required to delve into the biological mechanism of the formation and differentiation of PD-1+CD8+ T cells in GC. Besides, our usage of the minimal P value method to determine the cut-off values may have raised the difficulty of reproducibility. In addition, since our study demonstrated that intratumoral PD-1+CD8+ T cells is indicative of clinical outcomes, we encouraged future researchers to test whether PD-1+CD8+ T cells from peripheral blood could serve as a noninvasive biomarker. Also, molecular subtypes might be a confounding factor in survival or other analysis, considering the imbalanced distribution of PD-1+CD8+ T cells among different molecular subtypes. However, it was not adjusted for the lack of such information based on high-throughput mRNA transcriptome data in ZSHS cohort.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data and materials generated that are relevant to the results are included in this article. Other data are available from the corresponding author Prof. Xu upon reasonable request.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NCT, Lordick F. Gastric cancer. Lancet. 2020;396:635–48.
Songun I, Putter H, Kranenbarg EM-K, Sasako M, van de Velde CJH. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol. 2010;11:439–49.
Fuchs CS, Mayer RJ. Gastric carcinoma. N. Engl J Med. 1995;333:32–41.
Smyth EC, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D, et al. Gastric cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27:v38–v49.
Ajani JA, D’Amico TA, Almhanna K, Bentrem DJ, Chao J, Das P, et al. Gastric cancer, version 3.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14:1286–312.
Nishida T. Adjuvant therapy for gastric cancer after D2 gastrectomy. Lancet. 2012;379:291–2.
Sasako M, Sakuramoto S, Katai H, Kinoshita T, Furukawa H, Yamaguchi T, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol. 2011;29:4387–93.
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8.
Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9.
Derks S, de Klerk LK, Xu X, Fleitas T, Liu KX, Liu Y, et al. Characterizing diversity in the tumor-immune microenvironment of distinct subclasses of gastroesophageal adenocarcinomas. Ann Oncol. 2020;31:1011–20.
Cao Y, He H, Li R, Liu X, Chen Y, Qi Y, et al. Latency-associated peptide identifies immunoevasive subtype gastric cancer with poor prognosis and inferior chemotherapeutic responsiveness. Ann Surg. 2020. https://doi.org/10.1097/SLA.0000000000003833.
Tsujimoto H, Ono S, Ichikura T, Matsumoto Y, Yamamoto J, Hase K. Roles of inflammatory cytokines in the progression of gastric cancer: friends or foes? Gastric Cancer. 2010;13:212–21.
Zhang H, Li R, Cao Y, Gu Y, Lin C, Liu X, et al. Poor clinical outcomes and immunoevasive contexture in intratumoral IL-10-producing macrophages enriched gastric cancer patients. Ann Surg. 2020. https://doi.org/10.1097/SLA.0000000000004037.
Liu X, Cao Y, Li R, Gu Y, Chen Y, Qi Y, et al. Poor clinical outcomes of intratumoral dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin-positive macrophages associated with immune evasion in gastric cancer. Eur J Cancer. 2020;128:27–37.
** K, Cao Y, Gu Y, Fang H, Fei Y, Wang J, et al. Poor clinical outcomes and immunoevasive contexture in CXCL13+CD8+ T cells enriched gastric cancer patients. Oncoimmunology. 2021;10:1915560.
Gu Y, Chen Y, ** K, Cao Y, Liu X, Lv K, et al. Intratumoral CD103(+)CD4(+) T cell infiltration defines immunoevasive contexture and poor clinical outcomes in gastric cancer patients. Oncoimmunology. 2020;9:1844402.
Fei Y, Cao Y, Gu Y, Fang H, Chen Y, Wang J, et al. Intratumoral Foxp3(+)RORgammat(+) T cell infiltration determines poor prognosis and immunoevasive contexture in gastric cancer patients. Cancer Immunol Immunother. 2021. https://doi.org/10.1007/s00262-021-02950-3.
Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599–611.
Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34.
Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33:547–62.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80.
Kim HD, Song GW, Park S, Jung MK, Kim MH, Kang HJ, et al. Association between expression level of PD1 by tumor-infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology. 2018;155:1936–.e1917.
Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, et al. TCR repertoire diversity of peripheral PD-1(+)CD8(+) T cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol Res. 2020;8:146–54.
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007;104:3360–5.
Wu X, Zhang H, **ng Q, Cui J, Li J, Li Y, et al. PD-1(+) CD8(+) T cells are exhausted in tumours and functional in draining lymph nodes of colorectal cancer patients. Br J Cancer. 2014;111:1391–9.
Han HS, Jeong S, Kim H, Kim HD, Kim AR, Kwon M, et al. TOX-expressing terminally exhausted tumor-infiltrating CD8(+) T cells are reinvigorated by co-blockade of PD-1 and TIGIT in bladder cancer. Cancer Lett. 2021;499:137–47.
Shen Y, Teng Y, Lv Y, Zhao Y, Qiu Y, Chen W, et al. PD-1 does not mark tumor-infiltrating CD8+ T cell dysfunction in human gastric cancer. J Immunother Cancer 2020;8:e000422.
Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68:1764–73.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–56.
Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80.
Chakravarty D, Solit DB. Clinical cancer genomic profiling. Nat Rev Genet. 2021;22:483–501.
Nakamura Y, Kawazoe A, Lordick F, Janjigian YY, Shitara K. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: an emerging paradigm. Nat Rev Clin Oncol. 2021;18:473–87.
Lordick F, Shitara K, Janjigian YY. New agents on the horizon in gastric cancer. Ann Oncol. 2017;28:1767–75.
Acknowledgements
We thank Dr. Lingli Chen (Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai, China) and Dr. Yunyi Kong (Department of Pathology, Shanghai Cancer Center, Fudan University, Shanghai, China) for their excellent pathological technology help.
Funding
This study was funded by grants from National Natural Science Foundation of China (31770851, 81871926, 81871930, 81902402, 81902901, 81972219, 82003019, 82103313 and 82172694), Shanghai Rising-Star Program (22QA1401700) and Shanghai Sailing Program (18YF1404600, 19YF1407500, 21YF1407600). All the sponsors have no roles in the study design, in the collection, analysis and interpretation of data.
Author information
Authors and Affiliations
Contributions
KY, YG and PZ for the acquisition of data, analysis and interpretation of data, statistical analysis and drafting of the manuscript; HF, YC, JW, CL, H. Liu, HZ and HH for technical and material support; RL, JQ, H. Li and JX for study concept and design, analysis and interpretation of data, drafting of the manuscript, obtained funding and study supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Clinical Research Ethics Committee of Zhongshan Hospital, Fudan University, with the approval number Y2015-054. Written informed consent was obtained from each patient included, and this study was performed in accordance with the Declaration of Helsinki.
Consent to publish
Consent for publication was obtained from each author.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, K., Gu, Y., Zhang, P. et al. Intratumoral PD-1+CD8+ T cells associate poor clinical outcomes and adjuvant chemotherapeutic benefit in gastric cancer. Br J Cancer 127, 1709–1717 (2022). https://doi.org/10.1038/s41416-022-01939-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01939-8
- Springer Nature Limited
This article is cited by
-
Diffuse large B-cell lymphoma: the significance of CD8+ tumor-infiltrating lymphocytes exhaustion mediated by TIM3/Galectin-9 pathway
Journal of Translational Medicine (2024)
-
Circulating memory PD-1+CD8+ T cells and PD-1+CD8+T/PD-1+CD4+T cell ratio predict response and outcome to immunotherapy in advanced gastric cancer patients
Cancer Cell International (2023)
-
Clinical implications of aberrant PD-1 expression for acute leukemia prognosis
European Journal of Medical Research (2023)
-
Prognostic value of soluble PD-L1 and exosomal PD-L1 in advanced gastric cancer patients receiving systemic chemotherapy
Scientific Reports (2023)
-
Clinical relevance of PD-1 positive CD8 T-cells in gastric cancer
Gastric Cancer (2023)