
Abstract
Aberrant activation of EGFR represents a common event in non-small cell lung carcinoma (NSCLC) and activates various downstream signaling pathways. While EGFR activation of β-catenin signaling was previously reported, the mediating mechanism remains unclear. Our current study found that EGFR activation in NSCLC cells releases SHC-binging protein 1 (SHCBP1) from SHC adaptor protein 1 (SHC1), which subsequently translocates into the nucleus and directly promotes the transactivating activity of β-catenin, consequently resulting in development of NSCLC cell stemness and malignant progression. Furthermore, SHCBP1 promotes β-catenin activity through enhancing the CBP/β-catenin interaction, and most interestingly, a candidate drug that blocks the CBP/β-catenin binding effectively abrogates the aforementioned biological effects of SHCBP1. Clinically, SHCBP1 level in NSCLC tumors was found to inversely correlate with patient survival. Together, our study establishes a novel convergence between EGFR and β-catenin pathways and highlights a potential significance of SHCBP1 as a prognostic biomarker and a therapeutic target.
Similar content being viewed by others


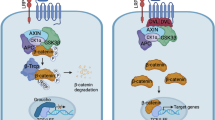
Introduction
Lung cancer is the most commonly diagnosed cancer type and a leading cause of cancer death globally. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases. Despite the availability of surgical therapy, radiotherapy, and chemotherapy, prognosis of NSCLC is still poor with overall five-year survival rate being as low as 15%, mainly due to development of resistance to chemo- and radiotherapy, postoperative recurrence and early metastasis [1,2,3,4,5,6]. Even though molecular targeted therapeutic drugs, e.g. EGFR tyrosine kinase inhibitors (TKIs), have shown encouraging efficacies on NSCLC patients in recent years, the vast majority of NSCLC patients who are initially sensitive to TKIs acquire TKI resistance and undergo relapse, metastasis, or other progressions ultimately [7, 8].
Cancer stem cells (CSCs) are subpopulations of malignant cells that possess the abilities to self-renew and differentiate within a tumor [9]. The biological properties of CSCs have been linked to tumor resistance to chemotherapy and radiation, post-treatment recurrence, and metastasis, and presumably, specific, effective CSC targeting strategies might suppress cancer relapse [10, 11]. Notably, while the molecular mechanism via which cancer cells acquire stemness and the acquired stemness is maintained remains to be understood, Wnt/β-catenin signaling has been evidently associated with the development of cellular stemness in both cancer and benign tissues [12, 13]. Canonically, activation of the Wnt/β-catenin pathway is initiated by binding of Wnt ligands to their transmembrane receptors, followed by sequestration of β-catenin in the cytoplasm away from the destined destruction complex so that β-catenin can enter the nucleus and activate transcription of its target genes, many of which have been found to contribute to the development of cellular stemess [14]. Of note, activation of β-catenin signaling has been well demonstrated in various cancer types, most of which is attributable to gene alterations of the key components of β-catenin signaling. Typically, in colorectal tumors, the vast majority (80–90%) of clinical cases contain frameshift or truncating mutations in APC, resulting in the loss of ability to binding β-catenin [15]. Mutations of AXIN, which also lead to disruption of the destruction complex, have been identified likewise. In addition, mutations of β-catenin phosphorylation sites and consequent abrogation of β-catenin phosphorylation have been found in melanoma, which leads to β-catenin accumulation in the nucleus and transcription activation of its target genes [16, 17]. In such a context, of great interest is the fact that while enhanced nuclear localization of β-catenin has been observed in NSCLC [18] and hyperactive Wnt/β-catenin signaling is associated with increased drug resistance and distant metastasis of NSCLC [19], the aforementioned mutations are rare in NSCLC [20]. Hence, the molecular mechanisms underlying the activation of the pro-stemness β-catenin signaling in NSCLC remain to be investigated.
Of note, activating mutations of EGFR are common in NSCLC. Previous reports have shown a positive correlation between the presence of activating EGFR mutations and activation of β-catenin signaling in NSCLC [21], and the convergences between these two pathways have been indicated at multiple subcellular levels [21,22,23,24,25]. Notably, EGFR signaling reportedly increases cytoplasmic accumulation of β-catenin and nuclear translocation by either promoting release of β-catenin from the cytoplasmic membrane or disrupting the β-catenin destruction complex [24,25,26,27, All tumor cell lines were obtained from the Cell Bank of Shanghai Institutes of Biological Sciences (Shanghai, China), Fu Erbo Biotechnology Co., Ltd (Guangzhou, China), or ATCC, and cultured as previously described [55]. Primary NLE cells were obtained and cultured in keratinocyte-SFM medium (KSFM) [55]. Authenticity of the cell lines was verified by short tandem repeat (STR) fingerprinting at the Medicine Laboratory of Forensic Medicine Department of Sun Yat-sen University (Guangzhou, China). Clinical tissue specimens were histopathologically diagnosed at the Sun Yat-Sen University Cancer Center from 2000 to 2004. The histological characterization and clinicopathologic staging of the cases were determined by following the standard provided in the current Union for International Cancer Control (UICC) Tumor-Node-Metastasis (TNM) classification. Each tumor and adjacent non-cancerous lung tissue pair was obtained according to our previous reports [55, 56]. Prior patients’ consents and approval from the Institutional Research Ethics Committee were obtained. The SHCBP1 expression plasmid was generated by PCR subcloning the human SHCBP1 coding sequence into the lentiviral transfer plasmid pSin-puro (Clontech, Palo Alto, CA) to generate plasmid pSin-SHCBP1. To deplete SHCBP1 expression, two human shRNA sequences (sh1: GCGATTCAGAGCCTATCAA; sh2: CCATAGTGATCCATTGTCT) were cloned into the pSuper-retro-puro plasmid to generate pSuper-retro-SHCBP1-sh1 and pSuper-retro-SHCBP1-sh2. Human EGF-coding sequence was subcloned into the lentiviral transfer plasmid to generate plasmid pSin-EGF. Retroviral and lentiviral production and infection were performed according to the manufacturer’s instructions. Total RNA extraction, reverse transcription, and real-time PCR were performed as described previously [55]. Primers were purchased from Invitrogen. The cDNA was acquired by using the GoScript™ Reverse Transcription Mix (Promega). All results were normalized for the expression of GAPDH, and relative quantification was calculated using the 2−△△CT formula. Lysates were prepared from 3 × 107 293T cells transduced with Flag-tagged SHCBP1 or vector using lysis buffer. Lysates were then incubated with FLAG affinity agarose (Sigma-Aldrich, St Louis, MO) overnight at 4 °C. Beads containing affinity-bound proteins were washed six times with immunoprecipitation wash buffer. Proteins were separated on SDS polyacrylamide gels stained with Coomassie blue, and all bands were subjected to mass spectrometry analysis. SHCBP1 and β-catenin protein purification was acquired by using immunoprecipitation and FLAG/HA competing peptides (MedChemExpress, Monmouth, NJ, USA) (see Supplementary Materials and Methods for details). Western blotting analysis was performed as described previously [55, 57]. The antibodies used are listed in Supplementary Information. Cells were seeded in triplicates in 24- or 48-well plates and allowed to settle for 24 h. Indicated plasmids plus 10 ng pRL-TK renilla plasmid was transfected into the cells using the Lipofectamine 3000 reagent (Invitrogen). Forty-eight hours after transfection, Dual-Luciferase reporter assays were performed according to the manufacturer’s protocol of Dual Luciferase Reporter Assay Kit (Promega, Madison, WI). The reporter plasmids containing wild-type (CCTTTGATC; TOPflash) or mutated (CCTTTGGCC; FOPflash) TCF/LEF DNA-binding sites were purchased from Upstate Biotechnology (Lake Placid, NY). NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Scientific ™ Pierce, USA) was used and the assays were performed according to the manufacturer’s instructions. Tumor spheres were cultured according to a previous report [58]. Cells were allowed to further grow for 10 days, and the numbers of spheres were microscopically counted. The sphere yields were calculated by the number of spheres derived from cells based on the initially plated 2500 cells. IHC assays in 207 NSCLC tissues were performed by using primary SHCBP1 antibody (Abgent, San Diego, CA) and quantified according to our previous report [59, 60]. The degree of immunostaining of indicated proteins was evaluated and scored by two independent observers as previously described, scoring both the proportions of positive staining tumor cells and the staining intensities. Scores representing the proportion of positively stained tumor cells was graded as: 0 (no positive tumor cells), 1 (<10%), 2 (10–50%), and 3 (>50%). The intensity of staining was determined as: 0 (no staining); 1 (weak staining = light yellow), 2 (moderate staining = yellow brown), and 3 (strong staining = brown). The staining index (SI) was calculated as the product of staining intensity × percentage of positive tumor cells, resulting in scores as 0, 1, 2, 3, 4, 6, and 9. Cutoff values for high- and low-expression of protein of interest were chosen based on a measurement of heterogeneity using the log-rank test with respect to overall survival. The optimal cutoff was identified as: the SI score of ≥4 was considered as high expression, and ≤3 as low expression. Female BALB/c-nu mice (5–6 weeks of age, 18–20 g) were purchased and housed in specific pathogen-free facilities on a 12-h light/dark cycle. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University. At least five nude mice per group were used to ensure the adequate power and each mouse with different weight was randomly allocated. ICG-001 or vehicle has begun to be used 7 days after tumor cell injections in animals. ICG-001 (50 mg/kg) or vehicle (PBS) were intraperitoneally injected every 3 days for 3 weeks as described previously [50, 61]. For bioluminescent imaging assay, 15 min prior to imaging, mice were injected intraperitoneally (i.p.) with 150 mg/kg luciferin. Following general anesthesia, images were taken and analyzed with Spectrum Living Image 4.0 software (Caliper Life Sciences). Tumor growth was monitored weekly by in vivo imaging and photon radiance measurement, and the final monitoring was performed after 4 or 6 weeks after the injection. Bioluminescent imaging of primary tumors and metastases was performed in a blinded manner. Sample size was determined by power analysis to achieve a minimum effect size of 0.5 with P < 0.05 and all sample sizes were appropriate for assumption of normal distribution. Variance within each group of data was estimated and was similar between compared groups. Data analysis was performed by two independent investigators who were blinded to the sample groups. All statistical analyses were performed using the SPSS 13.0 (IBM) statistical software package. The Kaplan–Meier method was used to establish survival curves. The statistical significance of various variables for survival was analyzed using the Cox proportional hazards model in the multivariate analysis. Correlation between SHCBP1 expression and T-, N-, M-classification was analyzed using chi-square test. Comparisons between groups were performed with a two-tailed paired Student’s t-test. In all cases, P < 0.05 was defined as statistically significant.Materials and methods
Cell cultures
Tumor specimens from patients
Plasmids and transfection
RNA extraction and real-time PCR
Immunoprecipitation and protein purification
Western blotting analysis
Luciferase reporter assay
Nuclear and cytoplasmic extraction
Primary sphere formation
IHC assays
Animal studies
Statistical analysis
References
Feld R, Rubinstein LV, Weisenberger TH. Sites of recurrence in resected stage I non-small-cell lung cancer: a guide for future studies. J Clin Oncol. 1984;2:1352–8.
Passlick B, Izbicki JR, Kubuschok B, Nathrath W, Thetter O, Pichlmeier U, et al. Immunohistochemical assessment of individual tumor cells in lymph nodes of patients with non-small-cell lung cancer. J Clin Oncol. 1994;12:1827–32.
Berns A. Stem cells for lung cancer? Cell. 2005;121:811–3.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64.
Le Pechoux C. Role of postoperative radiotherapy in resected non-small cell lung cancer: a reassessment based on new data. Oncologist. 2011;16:672–81.
To KK, Tong WS, Fu LW. Reversal of platinum drug resistance by the histone deacetylase inhibitor belinostat. Lung Cancer. 2017;103:58–65.
Sequist LV, Joshi VA, Janne PA, Muzikansky A, Fidias P, Meyerson M, et al. Response to treatment and survival of patients with non-small cell lung cancer undergoing somatic EGFR mutation testing. Oncologist. 2007;12:90–8.
Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30:433–40.
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512.
Ishizawa K, Rasheed ZA, Karisch R, Wang Q, Kowalski J, Susky E, et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell. 2010;7:279–82.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Sokol SY. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development. 2011;138:4341–50.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80.
Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90.
Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–2.
Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 2009;28:2163–72.
Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62.
Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1–10.
Suzuki M, Shigematsu H, Nakajima T, Kubo R, Motohashi S, Sekine Y, et al. Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin Cancer Res. 2007;13:6087–92.
Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27:466–80.
Tan X, Apte U, Micsenyi A, Kotsagrelos E, Luo JH, Ranganathan S, et al. Epidermal growth factor receptor: a novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology. 2005;129:285–302.
Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36:547–59.
Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515.
Roth JF, Shikama N, Henzen C, Desbaillets I, Lutz W, Marino S, et al. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J. 2003;22:5186–96.
Ma L, Zhang G, Miao XB, Deng XB, Wu Y, Liu Y. et al. Cancer stem-like cell properties are regulated by EGFR/AKT/beta-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013;280:2027–41.
Fang D, Hawke D, Zheng Y, **a Y, Meisenhelder J, Nika H, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9.
Ma L, Zhang G, Miao XB, Deng XB, Wu Y, Liu Y, et al. Cancer stem-like cell properties are regulated by EGFR/AKT/beta-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013;280:2027–41.
Casas-Selves M, Kim J, Zhang Z, Helfrich BA, Gao D, Porter CC, et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res. 2012;72:4154–64.
Sato S, Idogawa M, Honda K, Fujii G, Kawashima H, Takekuma K, et al. Beta-catenin interacts with the FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenterology. 2005;129:1225–36.
Park M, Lim JS, Lee HJ, Na K, Lee MJ, Kang CM, et al. Distinct protein expression profiles of solid-pseudopapillary neoplasms of the pancreas. J Proteome Res. 2015;14:3007–14.
Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499:166–71.
Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31:2714–36.
Yang W, **a Y, Ji H, Zheng Y, Liang J, Huang W et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22.
Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–31.
Takemaru KI, Moon RT. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J Cell Biol. 2000;149:249–54.
Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci USA. 2004;101:12682–7.
Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–34.
Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403.
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–62.
Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci USA. 2002;99:14789–94.
Schmandt R, Liu SK, McGlade CJ. Cloning and characterization of mPAL, a novel Shc SH2 domain-binding protein expressed in proliferating cells. Oncogene. 1999;18:1867–79.
Togashi Y, Hayashi H, Terashima M, de Velasco MA, Sakai K, Fujita Y, et al. Inhibition of beta-Catenin enhances the anticancer effect of irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer with a T790M mutation. J Thorac Oncol. 2015;10:93–101.
Hwang KE, Kwon SJ, Kim YS, Park DS, Kim BR, Yoon KH, et al. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp Cell Res. 2014;323:288–96.
Nakayama S, Sng N, Carretero J, Welner R, Hayashi Y, Yamamoto M, et al. beta-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 2014;74:5891–902.
Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, et al. Receptor tyrosine kinases activate canonical WNT/beta-catenin signaling via MAP kinase/LRP6 pathway and direct beta-catenin phosphorylation. PLoS ONE. 2012;7:e35826.
Zhang X, Zhu J, Li Y, Lin T, Siclari VA, Chandra A, et al. Epidermal growth factor receptor (EGFR) signaling regulates epiphyseal cartilage development through beta-catenin-dependent and -independent pathways. J Biol Chem. 2013;288:32229–40.
Wend P, Fang L, Zhu Q, Schipper JH, Loddenkemper C, Kosel F, et al. Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013;32:1977–89.
Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–78.
He K, Xu T, Xu Y, Ring A, Kahn M, Goldkorn A. Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem-like phenotype through modulation of the PI3K/Akt/beta-catenin/CBP pathway. Int J Cancer. 2014;134:43–54.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64.
Montembault E, Zhang W, Przewloka MR, Archambault V, Sevin EW, Laue ED et al. Nessun Dorma, a novel centralspindlin partner, is required for cytokinesis in Drosophila spermatocytes. J Cell Biol. 2010;191:1351–65.
Chen J, Lai F, Niswander L. The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev. 2012;26:803–15.
Yang Y, Liu L, Zhang Y, Guan H, Wu J, Zhu X, et al. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. Int J Cancer. 2014;135:1531–42.
Cai J, Wu J, Zhang H, Fang L, Huang Y, Yang Y et al. miR-186 downregulation correlates with poor survival in lung adenocarcinoma, where it interferes with cell-cycle regulation. Cancer Res. 2013;73:756–66.
Su S, Liao J, Liu J, Huang D, He C, Chen F, et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017;27:461–82.
Soeda A, Inagaki A, Oka N, Ikegame Y, Aoki H, Yoshimura S, et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283:10958–66.
Yang Y, Liu L, Cai J, Wu J, Guan H, Zhu X et al. DEPDC1B enhances migration and invasion of non-small cell lung cancer cells via activating Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 450: 899-905.
Lian C, Wu Z, Gao B, Peng Y, Liang A, Xu C, et al. Melatonin reversed tumor necrosis factor-alpha-inhibited osteogenesis of human mesenchymal stem cells by stabilizing SMAD1 protein. J Pineal Res. 2016;61:317–27.
Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, et al. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther. 2014;13:2303–14.
Acknowledgements
This work was supported by The National Key Research and Development Project of China (No. 2017YFA0505600, 2017YFA0106300); The Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 81621004); Guangzhou science and technology plan (No. 201803010039, 201804010057); The National Natural Science Foundation of China (No. 81572837, 81330058); Guangdong Natural Science Foundation (No. 2014A030313036); Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2014A030306023); Guangdong Te Zhi program youth science and technology talent of project (No. 2015TQ01R281); the Fundamental Research Funds for the Central Universities (No. 17ykpy06).
Author contributions
LL and YY participated in the design of the study, carried out experiments, analyzed data, and wrote a draft of the paper with the help of SL, TT, JC, JW, HG, XZ, and ZH. JL, ES, and MZ provided the associated clinical data from the subjects and provided technical assistance. ML conceived the ideas, designed and discussed experiments, supervised progress and extensively edited and communicated regarding the manuscript. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, L., Yang, Y., Liu, S. et al. EGF-induced nuclear localization of SHCBP1 activates β-catenin signaling and promotes cancer progression. Oncogene 38, 747–764 (2019). https://doi.org/10.1038/s41388-018-0473-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0473-z
- Springer Nature Limited