
Abstract
Smoking is a leading cause of preventable morbidity and mortality. Smoking is heritable, and genome-wide association studies (GWASs) of smoking behaviors have identified hundreds of significant loci. Most GWAS-identified variants are noncoding with unknown neurobiological effects. We used genome-wide genotype, DNA methylation, and RNA sequencing data in postmortem human nucleus accumbens (NAc) to identify cis-methylation/expression quantitative trait loci (meQTLs/eQTLs), investigate variant-by-cigarette smoking interactions across the genome, and overlay QTL evidence at smoking GWAS-identified loci to evaluate their regulatory potential. Active smokers (N = 52) and nonsmokers (N = 171) were defined based on cotinine biomarker levels and next-of-kin reporting. We simultaneously tested variant and variant-by-smoking interaction effects on methylation and expression, separately, adjusting for biological and technical covariates and correcting for multiple testing using a two-stage procedure. We found >2 million significant meQTL variants (padj < 0.05) corresponding to 41,695 unique CpGs. Results were largely driven by main effects, and five meQTLs, map** to NUDT12, FAM53B, RNF39, and ADRA1B, showed a significant interaction with smoking. We found 57,683 significant eQTL variants for 958 unique eGenes (padj < 0.05) and no smoking interactions. Colocalization analyses identified loci with smoking-associated GWAS variants that overlapped meQTLs/eQTLs, suggesting that these heritable factors may influence smoking behaviors through functional effects on methylation/expression. One locus containing MUSTN1 and ITIH4 colocalized across all data types (GWAS, meQTL, and eQTL). In this first genome-wide meQTL map in the human NAc, the enriched overlap with smoking GWAS-identified genetic loci provides evidence that gene regulation in the brain helps explain the neurobiology of smoking behaviors.
Similar content being viewed by others
Introduction
Genetic variants that act as quantitative trait loci (QTLs) for gene regulatory features, such as DNA methylation (DNAm) and RNA expression (RNAexp) levels, are pervasive across the genome [1,2,3,4,5] and are enriched among disease-associated loci [6,29]. DNAm was measured using an Illumina Human MethylationEPIC BeadChip. As described before [11], DNAm data processing and QC were conducted using the R package minfi [30]. DNAm β-values were calculated and used in the meQTL analyses, representing the percentage of DNAm at each CpG (ratio of methylated intensities relative to the total intensity). See Supplementary Materials for more details.
In addition, the DNAm processing, sample and probe exclusions, and modeling information are described extensively in the Supplementary Materials of Markunas et al. [11].
Following RNA extraction, samples were sequenced using paired-end 100 bp reads on an Illumina HiSeq3000 at LIBD [11, 31]. RNA-seq samples were pseudomapped using Salmon v1.1.0 [32]. Transcript quantifications were aggregated to the gene-level counts using tximport v1.12.3 [33], resulting in 60,240 GENCODE v34 genes. Samples were excluded based on RNA-seq quality metrics, low RNA integrity (RIN) score, discrepancies between self-reported sex and chromosome Y gene expression, discrepancies between RNA and DNA derived genotype data, and missing genotype data. Sample-level QC removed 36 samples, resulting in a post-QC RNA-seq sample size of 203. Lowly expressed genes were then removed using the exclusion criteria of ≥90% of samples with ≤10 gene counts or ≤1 transcripts per million value. For eQTL map**, GRCh37 human genome reference coordinates were used for all gene annotations to align with the genotype data genome build. In total, 16,274 genes were considered for eQTL map**. See Supplementary Materials for more details.
Methylation and expression quantitative trait loci (meQTL/eQTL) map**
We performed cis-meQTL map** using imputed genetic variants with an MAF > 0.05 and DNAm intensity β-values of probes proximal, within 500 kilobases (kb) up or downstream, to these variants. Four different meQTL map** regression models were fit: (1) a “baseline” model to test for association between genetic variants and DNAm β-values across both smoking cases and controls; this model included age at death, sex, estimated non-neuronal cell-type proportion [34], principal component 1 (PC1) for DNAm array negative control probes, PC1 for imputed genotypes, and PC2 for imputed genotypes as covariates (see Supplementary Methods for model selection details); (2) a smoking cases–only model similar to the baseline model; (3) a smoking controls–only model similar to the baseline model; and (4) an interaction model to test for associations of a genetic variant-by-smoking status interaction with DNAm β-values. All models used rank-inverse normal transformed (RINT) DNAm β-values. The smoking cases–only and controls–only models are needed to generate summary statistics used to conduct stratified two degrees-of-freedom (2DF) tests. The stratified 2DF test jointly tests for genetic variant main effects and genetic variant-by-smoking status interaction effects [35]. Additional details are provided in the Supplementary Methods.
A similar framework was applied for cis-eQTLs. Baseline, smoking cases–only, smoking controls–only, and interaction eQTL models were fit for each imputed genetic variant and NAc expression levels of genes proximal (within 500 kb of gene body) to these variants. Counts for a given gene underwent median-of-ratios normalization [36] and RINT to obtain values used to represent the gene’s expression level. The covariates included in each model were age at death, sex, exon map** rate, ribosomal RNA map** rate, PC1 for imputed genotypes, PC2 for imputed genotypes, and four latent variables estimated by PEER v1.3 [37] to account for additional unmeasured sources of confounding (see Supplementary Methods).
To account for multiple hypothesis testing in the meQTL/eQTL models and stratified 2DF tests, p values were corrected using a conversative two-stage approach to mitigate inflation associated with single-stage approaches when applied to QTL map** [38]. This hierarchical approach first accounts for association tests across variants for a given DNAm probe/gene, then accounts for tests across all probes/genes. In the first stage, all nominal p values for a given probe/gene were adjusted using EigenMT [39]. In the second stage, EigenMT adjusted p values were further corrected by the number of probes/genes tested. Any initial two-stage adjusted p value smaller than a Bonferroni corrected p value was assigned the latter p value as the final two-stage adjusted p value. This ensures that the two-stage adjustment stringency does not exceed the family-wise error rate control of Bonferroni correction. A two-stage adjusted p value cutoff of 0.05 was used to identify statistically significant QTLs.
Gene Set Enrichment Analysis (GSEA) for eGenes and meQTL CpGs
We used the GSEAPreranked tool from GSEA v4.3.2 [40] to test for enrichment of smoking-associated eGenes and meQTL CpGs in the MSigDB v2023.1.Hs gene set collections C2 canonical pathways and C5 Gene Ontology gene sets. Only gene sets with 15–500 genes were included. CpGs were assigned to genes using the Infinium MethylationEPIC v1.0 B5 Manifest, resulting in 11,772 CpGs from the interaction meQTL map** model with at least one assigned gene. Each gene was ranked using the largest −log10-transformed, smoking interaction p value for a given gene from QTL map**. Weighted Kolmogorov–Smirnov–like statistics were computed for the enrichment scores, and p values were determined using 1000 gene set permutations. Significant pathways were selected based on false discovery rate (FDR) < 0.10, which was selected to maximize biological discovery and yet be more stringent than the default threshold of 0.25.
QTL enrichment testing for GSCAN-identified genetic variants
We conducted variant-based enrichment testing to assess whether genome-wide significant GSCAN loci were enriched for meQTLs or eQTLs. We obtained GSCAN summary statistics from the University of Minnesota’s Data Repository for U of M (https://doi.org/10.13020/3b1n-ff32), focusing on GSCAN’s GWAS results from 2019 to capture genetic loci with variants that commonly occur and have the largest effect sizes on smoking: N up to 1.2 million individuals, depending on the smoking trait analyzed [19]. For meQTL enrichment analysis, we compared the p value distributions from stratified 2DF tests (i.e., stratified 2DF meQTL map**) between GSCAN variants and a set of randomly matched variants. The random matched variant set was designed to be 10 times the size of the GSCAN variant set. The GSCAN variant set included linkage disequilibrium (LD)-pruned, genome-wide significant variants reported by GSCAN, across all four smoking traits, that were also available in our meQTL map** (361 variants, each representing an independent significant locus). The matched variant set included 3,600 LD-pruned variants selected using SNPsnap [41]. For variants with multiple stratified 2DF p values (i.e., proximal to multiple CpG sites), only the smallest p value was retained. The meQTL stratified 2DF p value distributions for the GSCAN and matched variant sets were tested for equality using a two-sided Kolmogorov–Smirnov test. The eQTL enrichment analysis followed the same procedure as the meQTL enrichment analysis. The GSCAN variant set included 305 variants because the overlap with variants from eQTL map** differed from meQTL map**. The SNPsnap-constructed, matched variant set included 3050 (305 × 10) variants.
Colocalization between GSCAN smoking GWAS and meQTL/eQTL map**s
We tested whether meQTL or eQTL signals from the baseline model QTL map** colocalized with GSCAN loci for smoking initiation, age at initiation, cigarettes per day, and cessation using the coloc v5.1.0 R package. We describe this analysis for meQTLs, but an equivalent framework was applied for eQTLs. All DNAm probes with an meQTL map** cis-window that overlapped with a GSCAN locus were considered for colocalization. For a given probe, the colocalization test region spanned all genetic variants that were (1) included in the meQTL map** for the probe and (2) tested in the GSCAN GWAS (Fig. S1). Summary statistics from GSCAN and the baseline model meQTL map** were used. Significant colocalization required a coloc posterior probability >0.8 for hypothesis 4 (both traits are associated and share a single causal variant) and a two-stage, adjusted p < 0.05 in the baseline meQTL analysis (Supplementary Materials).
For GSCAN loci that showed evidence of colocalization, HyPrColoc was applied to assess whether these colocalizations resulted from the same region of the locus [42]. Each HyPrColoc test included only one GSCAN trait, eGene, and CpG. For each HyPrColoc test, a genetic variant was included as input only if it had summary statistics available from the baseline model me/eQTL map** and GSCAN GWAS for the smoking trait. Significant colocalization was defined as a GSCAN trait–eGene–CpG triplet having posterior probability >0.8.
Results
Overview
Of the available 239 decedents, 201 RNA-seq samples and 220 DNAm samples, with genotype and smoking data, remained following QC, including 198 samples in both datasets (intersection) and 223 samples (52 cases and 171 controls) with either RNA-seq or DNAm data (union) (Table 1). Of the cases, 50% had African ancestry (AA) and 50% had European ancestry (EA) based on next-of-kin report and genotype confirmation. The manner of death differed slightly among cases and controls, but age, sex, and postmortem interval were similar (Table 1).
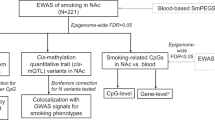
We generated single data-type QTL maps. Then each QTL type independently underwent colocalization analysis with GSCAN GWAS summary statistics, and joint colocalization analyses across data types were performed for significant me/eQTLs (Fig. 1).

QTL analyses were conducted using DNA methylation, gene expression, and genotype data and three different QTL models to produce genome-wide cis-eQTL and cis-meQTL maps for nucleus accumbens. Two-stage multiple testing correction was applied to these QTL maps to identify significant QTLs that underwent further annotation. QTL maps were also integrated with GSCAN GWAS summary statistics to perform genetic variant enrichment testing and colocalization analyses. Smoking interaction QTL maps were used in conjunction with the Molecular Signatures Database to conduct gene set enrichment analysis and identify biological pathways enriched for CpGs/genes associated with smoking status and genotype.
Genome-wide cis-meQTL maps
DNAm in NAc and genotype data were available for 52 smoking cases (26 EA, 26 AA) and 168 smoking controls (75 EA, 93 AA). In all, 11,206,899 variants were used in the initial analysis and 784,843 CpGs, resulting in 1,748,985,510 meQTL tests. After applying two-stage multiple testing correction, we identified 2,552,641 significant meQTL variants targeting 51,315 unique CpGs (Table S1) -which is restricted to the lead variant due to size; full results are available at synapse.org, https://doi.org/10.7303/syn50996324).
To identify the most robust signals, we performed post hoc filtering, kee** only meQTLs where the top variant had a minor allele frequency (MAF) ≥ 0.05 and missingness ≤0.10 in both ancestries, leaving 41,695 unique CpGs. The top five most significant meQTLs include CpGs that map to PITRM1, KDM3B, ARID1B, or MTL5 (Table S1).
Genome-wide cis-eQTL maps
Gene expression (RNA-seq) in NAc and genotype data were available for 47 smoking cases (24 EA, 23 AA) and 156 smoking controls (72 EA, 84 AA). These data were previously used to map eQTLs agnostic to the smoking phenotype [5]. In the present study accounting for variant-by-smoking interaction, we identified 83,095 significant eQTL variants from 1050 eGenes after multiple testing correction. Of these, 57,683 eQTL variants targeting 958 unique eGenes remained after post hoc filtering was applied to identify the most robust signals, kee** only eQTLs where the top variant had an MAF ≥ 0.05 and missingness ≤0.10 in both ancestries (Table S2). All significant results are shown in Table S2. Table S3 is filtered to the lead variant for each significant eGene (N = 1050). The 10 most significant eGenes were RPL9, ZSWIM7, GATD3B, RPS28, XRRA1, TMEM161B-AS1, CUTALP, NIPBL-DT, ZNF718, and SPATA7 (Table S3). Full results are available at synapse.org (https://doi.org/10.7303/syn50996324).
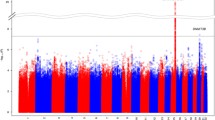
Both meQTLs and eQTLs were pervasive throughout the genome (Fig. 2). When we compared the eQTL to meQTL results, 562/1050 (54%) significant eGenes overlapped genes annotated to significant meQTL CpGs. Additionally, data from 655 (62%) robust eGenes (after MAF and missingness filtering) identified in our analysis were available in the NAc GTEx data. Of these 655, 509 (78%) met a Bonferroni correction significance threshold (0.05/655 = 7.6e-5) (Table S3, GTEx column: Nominal p-value). Thus, we observed a high correlation when comparing NAc eQTL results between our study and GTEx.

Stratified 2DF QTL map** −log10 nominal p values for significant (2DF adjusted p ≤ 0.05) genetic variant–CpG probe (top panel) and genetic variant-gene associations tests (bottom panel) are displayed as a function of genome position (x-axis). Genomic locations of significant GSCAN GWAS loci for four smoking phenotypes are denoted by the bars in the middle panel.
Smoking interaction effects with meQTLs/eQTLs
To identify meQTLs/eQTLs that differed by smoking, we compared the 2DF QTL test results with those of the baseline models and interaction models. The meQTLs were primarily driven by main effects, with few showing evidence for interaction. Of the 41,695 significant unique CpGs, only five demonstrated strong evidence of a smoking interaction (Table 2, Fig. S2) based on Bonferroni correction (significance threshold of 0.05/41,695 = 1.2e-6). No eQTLs showed evidence of an interaction (significance threshold of 0.05/877 unique eGenes=5.7e-5) after filtering by MAF and missingness.
Because use of stringent significance thresholds may miss subtle smoking interactions with individual variants, we performed GSEA using genes ranked based on QTL–smoking interaction p values to identify QTL-enriched biological processes and pathways that may be altered by smoking. For meQTL-smoking interactions, five pathways related to the synaptic cleft (neurotransmission site between pre- and postsynaptic membranes) were implicated. For eQTL-smoking interactions, cell cycle and wound response processes (cellular changes resulting from a stimulus indicated damage to an organism) were enriched (Table S4).
GWAS-identified variants that exert QTL effects on their target genes
To test the overlap** evidence of GWAS-identified variants as QTLs, we performed an enrichment analysis using a two-sample Kolmogorov–Smirnov test that compared NAc meQTL and eQTL p value distributions at GSCAN significant variants with a random set of variants. We found that GSCAN variants were significantly enriched for meQTLs (p = 0.005) but not for eQTLs (p = 0.3). This aligns with our observation that more meQTLs than eQTLs overlap GSCAN variants (Tables S5–S7). For comparison, using the same test with NAc eQTLs from GTEx, we found an enrichment p = 0.07.
Smoking meQTL, eQTL, and GWAS colocalization
We performed colocalization analyses of GSCAN’s GWAS results with our QTL maps to characterize heritable components of smoking that exert QTL effects. First, we performed pair-wise analyses (meQTL + GWAS, eQTL + GWAS), starting with GSCAN significant loci. Because a single locus may include more than one unique CpG or eGene, many colocalization analyses were performed per region. In general, we observed more colocalization of meQTLs than eQTLs with the GWAS loci (Tables S8–S10). Four genome-wide significant CpGs colocalized across two phenotypes (cigarettes per day and age of initiation): cg12293539 at MAML3 and cg00622170, cg11254171, and cg18236429 at the NOP14/NOP14-AS1 locus.
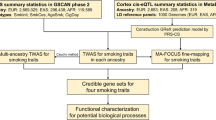
Next, we performed colocalization using a method (HyPrColoc) that can incorporate meQTLs, eQTLs, and smoking GWAS in a single analysis. Focusing on GSCAN GWAS loci that colocalized with both a significant eQTL and meQTL, we confirmed the GWAS–eQTL–meQTL colocalizing region at the GSCAN smoking initiation locus chr3: 52386605–54266212. Three variant–CpG–eGene combinations had significant colocalization, all involving ENSG00000243696 (predicted read-through of MUSTN1-ITIH4) as the eGene with (1) rs6445538 and cg25643088, (2) rs6445538 and cg19713033, and (3) rs4687672 and cg23815702 (Fig. 3A). The C allele of rs6445538 and the A allele of rs4687672 are associated with increased smoking initiation with very modest effect sizes: 0.023 for both [19, 43]. These alleles are associated with increased ENSG00000243696 gene expression (Table S2: Column: βbaseline), increased DNA methylation at cg25643088 and cg23815702, and decreased DNA methylation at cg19713033 (Fig. 3B, Table S1: Column: βbaseline).

A −log10 nominal p values for associations between genetic variants and smoking initiation, expression of three CpGs, and gene expression for the eGene identified by HyPrColoc with colocalization probability >0.8. Only genetic variants that overlapped across summary statistics for GSCAN, meQTL map**, and eQTL map** are plotted. Locations of genes included in the eQTL map** are annotated in the bottom of the panel. B Genotype-by-CpG plots for the CpGs in Fig. 3A showing CpG probe intensity levels partitioned by genotype.
Discussion
Cigarette smoking remains highly prevalent and a leading cause of death globally, despite decades of research into the health consequences and public health campaigns to curb smoking [44, 45]. Addiction to cigarette smoking is a complex, multi-stage process involving a neuronal rewards system that includes the NAc region of the brain [21, 46]. The NAc is known to have a role in cognitive processing of motivation, reward, and reinforcement, which are essential to the first stage of addiction (binge/intoxication) [22]. This study employed methods to better understand the functional effects of heritable factors that influence smoking behaviors.
QTLs are pervasive throughout the genome and provide valuable insight into tissue-specific gene regulation. The GTEx project, widely used for exploring eQTLs, recently released a large-scale meQTL dataset encompassing nine human tissues [47]. However, this dataset did not capture brain tissues. To our knowledge, ours is the first genome-wide meQTL map in human NAc and is shared publicly as a new resource for the scientific community.
We found few significant variant-by-smoking interaction effects and conclude that most QTLs in the NAc may not differ by smoking. While some interactions with small effect sizes may exist, they may require larger sample sizes to detect. We investigated this possibility using a pathway analysis ranked by evidence of smoking interactions, and we identified pathways related to cellular damage, cell cycle, and the synaptic cleft, where the nicotinic acetylcholine receptors play an important role in regulating neurotransmission [48]. Five individual meQTLs differed significantly between smokers and nonsmokers, including Nudix Hydrolase 12 (NUDT12), Family with sequence similarity 53 member B (FAM53B), and Ring Finger Protein 39 (RNF39). NUDT12 plays a role in nicotinate and nicotinamide metabolism [49]. Interestingly, NUDT12 was identified in a transcriptome analysis of neurons following chronic nicotine exposure [50] and lies within a QTL interval for nicotine sensitivity in mouse studies [51, 52]. FAM53B has been associated with cocaine dependence [53]. Differential DNAm at FAM53B has been observed in COPD cases compared with controls [54]. RNF39 was found to be differentially methylated in a study of marijuana use [55] and other smoking-related DNAm changes [56, 57]. Together, these represent biologically plausible genes whereby smoking may alter genetically driven gene regulation in NAc.
We employed colocalization analyses [5] to identify trait-associated variants that act as regulators of DNAm or gene expression. We generally observed more overlap between the GWAS-associated variants and meQTLs than eQTLs.The weaker overlap of GWAS variants and eQTLs has been noted elsewhere [58], and this pattern was also supported by our enrichment analyses, and may relate to the magnitude of meQTL tests. However, the same pattern held when we looked only at QTLs that survived genome-wide multiple testing correction, whereby declaring meQTLs as statistically significant was based on a more stringent threshold than eQTLs and is consistent with studies of the prefrontal cortex [59], blood [60], and other tissues [47].
One genomic region showed robust evidence of colocalization with all three data types, highlighting novel functional evidence where changes to DNAm and gene expression may help explain the neurobiology underlying a smoking initiation associated heritable factor. The primary gene indicated is a predicted read-through of the Musculoskeletal, Embryonic Nuclear Protein 1 (MUSTN1) and inter-alpha-trypsin inhibitor, heavy chain 4 (ITIH4) genes. Little is known about the read-through transcript. MUSTN1 is known to have a role in skeletal muscle homeostasis, chondrocyte differentiation, and limb morphogenesis [61]. Schizophrenia-associated variants in ITIH4 have been shown to regulate expression of ITIH4 in the prefrontal cortex [62], and variants in ITIH4 is a biomarker for COPD [63, 64]. MUSTN1 and ITIH4 are expressed at low levels in the brain and moderately expressed in skeletal muscle [1, 65, 66]. There is evidence that changes to skeletal muscle homeostasis can influence the physiology of the brain [67, 68]. However, we did not detect a significant eQTL for MUSTN1 or ITIH4 in our eQTL analysis, indicating that the MUSTN1-ITIH4 read-through may warrant further functional interrogation to decipher how its role may differ from MUSTN1 and ITIH4.
This study has limitations to consider in interpreting the findings. First, because this study utilized an understudied brain tissue collected from decedents with a unique set of multi-omics data types and smoking status, sample availability was limited to 52 smoking cases and 171 smoking controls. This constrained sample size may have limited our ability to identify interaction effects, and it limited interrogation of ancestry- or sex-specific effects and extension into independent replication datasets. Also, QTL map** results in millions of tests, increasing the chance of type I error. We accounted for multiple testing using a two-stage correction strategy designed for this type of study [39]; however, the possibility of type I error remains. Furthermore, despite the concordance between eGenes identified from eQTL map** and those reported by GTEx, the two-stage correction strategy we applied has reduced sensitivity to detect eQTL relative to the single-stage FDR correction approach applied by GTEx, resulting in a lower number of reported eGenes in our study despite comparable sample size.
Finally, the QTL–GWAS colocalization was based on the first GSCAN meta-analysis, including up to 1.2 million individuals [19]. GSCAN recently released an updated meta-analysis with >3 million individuals [20]. Although comparison with the updated GSCAN meta-analysis might result in the identification of additional QTL–GWAS colocalization signals, given the sample size available with multi-omics data in our postmortem human brain study, we would have less statistical power to detect colocalization with the lower MAF variants in GSCAN2, and these variants are not well captured by our imputed genotype data. The present study focused on genetic loci with common variants with the largest effect sizes on smoking.
This study had several strengths. It is the first to provide a genome-wide meQTL map in human NAc, a relatively understudied brain tissue with an important role in the addiction cycle. Case definitions were carefully established based on corroborating evidence from several sources, including blood- and brain-based toxicology screens with confirmation by next-of-kin reports. Therefore, misclassification is unlikely. We used the 2DF test, which achieves power similar to that of a standard 1DF test when no interaction is present and simultaneously improves power when interaction effects are present [35]. We used conservative thresholds to identify the most robust signals, and the full results are provided for researchers to explore additional signals. To our knowledge, the present study represents the first large-scale formal testing of colocalization for GSCAN-identified loci with QTLs in human brain, identifying novel target genes and providing insight into the neurobiological function of smoking-associated heritable factors in relation to both DNAm and gene expression.
Overall, this multi-ancestry, multi-omics study of decedents with smoking status known and accounted for provides a unique resource for interrogating regions across the genome for their influence on gene regulation, cigarette smoking behaviors, and other complex conditions involving the NAc. Future studies may use these data to compare QTLs across other brain tissues to gain insights into tissue-specific regulation and to further investigate the neurobiology underlying other disease processes.
References
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30.
Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53:1300–10.
Perzel Mandell KA, Eagles NJ, Wilton R, Price AJ, Semick SA, Collado-Torres L, et al. Genome-wide sequencing-based identification of methylation quantitative trait loci and their role in schizophrenia risk. Nat Commun. 2021;12:5251.
Min JL, Hemani G, Hannon E, Dekkers KF, Castillo-Fernandez J, Luijk R, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53:1311–21.
Chen F, Wang X, Jang SK, Quach BC, Weissenkampen JD, Khunsriraksakul C, et al. Multi-ancestry transcriptome-wide association analyses yield insights into tobacco use biology and drug repurposing. Nat Genet. 2023;55:291–300.
Markunas CA, Johnson EO, Hancock DB. Comprehensive evaluation of disease- and trait-specific enrichment for eight functional elements among GWAS-identified variants. Hum Genet. 2017;136:911–9.
Gamazon ER, Segre AV, van de Bunt M, Wen X, ** HS, Hormozdiari F, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018;50:956–67.
Barbeira AN, Bonazzola R, Gamazon ER, Liang Y, Park Y, Kim-Hellmuth S, et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021;22:49.
Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, et al. The impact of sex on gene expression across human tissues. Science. 2020;369:eaba3066.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–47.
Markunas CA, Semick SA, Quach BC, Tao R, Deep-Soboslay A, Carnes MU, et al. Genome-wide DNA methylation differences in nucleus accumbens of smokers vs. nonsmokers. Neuropsychopharmacology. 2021;46:554–60.
Semick SA, Collado-Torres L, Markunas CA, Shin JH, Deep-Soboslay A, Tao R, et al. Developmental effects of maternal smoking during pregnancy on the human frontal cortex transcriptome. Mol Psychiatry. 2020;25:3267–77.
Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464.
**ong X, Hou L, Park YP, Molinie B, GTEx Consortium, Gregory RI, et al. Genetic drivers of m(6)A methylation in human brain, lung, heart and muscle. Nat Genet. 2021;53:1156–65.
Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20:1418–26.
Hancock DB, Wang JC, Gaddis NC, Levy JL, Saccone NL, Stitzel JA, et al. A multiancestry study identifies novel genetic associations with CHRNA5 methylation in human brain and risk of nicotine dependence. Hum Mol Genet. 2015;24:5940–54.
Wang JC, Spiegel N, Bertelsen S, Le N, McKenna N, Budde JP, et al. Cis-regulatory variants affect CHRNA5 mRNA expression in populations of African and European ancestry. PLOS ONE. 2013;8:e80204.
Hancock DB, Guo Y, Reginsson GW, Gaddis NC, Lutz SM, Sherva R, et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol Psychiatry. 2018;23:1911–9.
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–44.
Saunders GRB, Wang X, Chen F, Jang SK, Liu M, Wang C, et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature. 2022;612:720–4.
Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3:760–73.
Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007;30:220–7.
BrainSeq: A Human Brain Genomics Consortium. BrainSeq: neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron. 2015;88:1078–83.
Avila-Tang E, Al-Delaimy WK, Ashley DL, Benowitz N, Bernert JT, Kim S, et al. Assessing secondhand smoke using biological markers. Tob Control. 2013;22:164–71.
Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case-control association studies. Nat Protoc. 2010;5:1564–73.
Delaneau O, Howie B, Cox AJ, Zagury JF, Marchini J. Haplotype estimation using sequencing reads. Am J Hum Genet. 2013;93:687–96.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13.
Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90:260–72.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Map** DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci. 2018;21:1117–25.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015;4:1521.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 2012;13:86.
Aschard H, Hancock DB, London SJ, Kraft P. Genome-wide meta-analysis of joint tests for genetic and gene-environment interaction effects. Hum Hered. 2010;70:292–300.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7:500–7.
Huang QQ, Ritchie SC, Brozynska M, Inouye M. Power, false discovery rate and Winner’s Curse in eQTL studies. Nucleic Acids Res. 2018;46:e133.
Davis JR, Fresard L, Knowles DA, Pala M, Bustamante CD, Battle A, et al. An efficient multiple-testing adjustment for eQTL studies that accounts for linkage disequilibrium between variants. Am J Hum Genet. 2016;98:216–24.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Pers TH, Timshel P, Hirschhorn JN. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–20.
Foley CN, Staley JR, Breen PG, Sun BB, Kirk PDW, Burgess S, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun. 2021;12:764.
PheWeb page for the GWAS & Sequencing Consortium of Alcohol and Nicotine (GSCAN). https://gscan.psych.umn.edu/, Accessed 3 April 2024.
Cornelius ME, Wang TW, Jamal A, Loretan CG, Neff LJ. Tobacco product use among adults – United States, 2019. MMWR Morb Mortal Wkly Rep. 2020;69:1736–42.
World Health Organization. WHO report on the global tobacco epidemic, 2017: monitoring tobacco use and prevention policies. World Health Organization, 2017.
Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38.
Oliva M, Demanelis K, Lu Y, Chernoff M, Jasmine F, Ahsan H, et al. DNA methylation QTL map** across diverse human tissues provides molecular links between genetic variation and complex traits. Nat Genet. 2023;55:112–22.
McKay BE, Placzek AN, Dani JA. Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharm. 2007;74:1120–33.
Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, et al. Genome-wide association study of smoking behaviours in patients with COPD. Thorax. 2011;66:894–902.
Yang J, Liu AY, Tang B, Luo D, Lai YJ, Zhu BL, et al. Chronic nicotine differentially affects murine transcriptome profiling in isolated cortical interneurons and pyramidal neurons. BMC Genom. 2017;18:194.
Gill KJ, Boyle AE. Genetic basis for the psychostimulant effects of nicotine: a quantitative trait locus analysis in AcB/BcA recombinant congenic mice. Genes Brain Behav. 2005;4:401–11.
Baker EJ, Jay JJ, Bubier JA, Langston MA, Chesler EJ. GeneWeaver: a web-based system for integrative functional genomics. Nucleic Acids Res. 2012;40:D1067–76.
Gelernter J, Sherva R, Koesterer R, Almasy L, Zhao H, Kranzler HR, et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry. 2014;19:717–23.
Lee MK, Hong Y, Kim SY, London SJ, Kim WJ. DNA methylation and smoking in Korean adults: epigenome-wide association study. Clin Epigenet. 2016;8:103.
Nannini DR, Zheng Y, Joyce BT, Kim K, Gao T, Wang J, et al. Genome-wide DNA methylation association study of recent and cumulative marijuana use in middle aged adults. Mol Psychiatry. 2023;28:2572–82.
Fuemmeler BF, Dozmorov MG, Do EK, Zhang JJ, Grenier C, Huang Z, et al. DNA methylation in babies born to nonsmoking mothers exposed to secondhand smoke during pregnancy: an epigenome-wide association study. Environ Health Perspect. 2021;129:57010.
Shorey-Kendrick LE, McEvoy CT, O’Sullivan SM, Milner K, Vuylsteke B, Tepper RS, et al. Impact of vitamin C supplementation on placental DNA methylation changes related to maternal smoking: association with gene expression and respiratory outcomes. Clin Epigenet. 2021;13:177.
Mostafavi H, Spence JP, Naqvi S, Pritchard JK. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat Genet. 2023;55:1866–75.
Lin H, Wang F, Rosato AJ, Farrer LA, Henderson DC, Zhang H. Prefrontal cortex eQTLs/mQTLs enriched in genetic variants associated with alcohol use disorder and other diseases. Epigenomics. 2020;12:789–800.
Pierce BL, Tong L, Argos M, Demanelis K, Jasmine F, Rakibuz-Zaman M, et al. Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms. Nat Commun. 2018;9:804.
Hadjiargyrou M. Mustn1: a developmentally regulated pan-musculoskeletal cell marker and regulatory gene. Int J Mol Sci. 2018;19:206.
Ohi K, Shimada T, Nitta Y, Kihara H, Okubo H, Uehara T, et al. Schizophrenia risk variants in ITIH4 and CALN1 regulate gene expression in the dorsolateral prefrontal cortex. Psychiatr Genet. 2016;26:142–3.
Bandow JE, Baker JD, Berth M, Painter C, Sepulveda OJ, Clark KA, et al. Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies–COPD biomarker discovery study. Proteomics. 2008;8:3030–41.
Lee KY, Feng PH, Ho SC, Chuang KJ, Chen TT, Su CL, et al. Inter-alpha-trypsin inhibitor heavy chain 4: a novel biomarker for environmental exposure to particulate air pollution in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:831–41.
The Human Protein Atlas. Accessed 21 May 2024 from https://www.proteinatlas.org.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Delezie J, Handschin C. Endocrine crosstalk between skeletal muscle and the brain. Front Neurol. 2018;9:698.
Isaac AR, Lima-Filho RAS, Lourenco MV. How does the skeletal muscle communicate with the brain in health and disease? Neuropharmacology. 2021;197:108744.
Acknowledgements
We gratefully thank the families who donated tissue to make this research possible. We also thank the Office of the Chief Medical Examiner of the State of Maryland, the Department of Pathology at Western Michigan University Homer Stryker MD School of Medicine, the University of North Dakota School of Medicine and Health Sciences’ Department of Pathology, the County of Santa Clara Medical Examiner-Coroner Office, and the National Institute of Mental Health (NIMH) Intramural Research Program for their collaboration on tissue collection.
Funding
This work was supported by the National Institute on Drug Abuse (NIDA) grants R01 DA042090 and R01 DA051913 to DBH and R21 DA051921 to HW.
Author information
Authors and Affiliations
Contributions
Author contributions are categorized using the CRediT taxonomy (credit.niso.org). Megan Ulmer Carnes: Formal analysis, methodology, visualization, writing – original draft, writing – review & editing. Bryan C. Quach: Formal analysis, methodology, software, visualization, writing – original draft, writing – review & editing. Linran Zhou: Formal analysis, visualization, writing – original draft, writing – review & editing. Shizhong Han: Resources, writing – review & editing. Ran Tao: Writing – review & editing. Meisha Mandal: Formal analysis, writing – review & editing. Amy Deep-Soboslay: Resources, writing – review & editing. Jesse A. Marks: Formal analysis, software, visualization, writing – original draft, writing – review & editing. Grier P. Page: Methodology, writing – review & editing. Brion S. Maher: Writing – review & editing. Andrew E. Jaffe: Resources, software, writing – review & editing. Hyejung Won: Resources, funding acquisition, writing – review & editing. Laura J. Bierut: Conceptualization, funding acquisition, writing – review & editing. Thomas M. Hyde: Resources, funding acquisition, writing – review & editing. Joel E. Kleinman: Resources, funding acquisition, writing – review & editing. Eric O. Johnson: Conceptualization, funding acquisition, writing – review & editing. Dana B. Hancock: Conceptualization, funding acquisition, project administration, supervision, writing – review & editing.
Corresponding author
Ethics declarations
COMPETING INTERESTS
MUC: None. BCQ: None. LZ: None. SH: None. RT: None. MM: None. AD-S: None. JAM: None. GPP: None. BSM: None. AEJ: AEJ is currently an employee and shareholder of Neumora Therapeutics; this connection is unrelated to the contents of this manuscript. HW: None. LJB: LJB is listed as an inventor on U.S. Patent 8,080,371,“Markers for Addiction,” covering the use of specific genetic variants in determining the diagnosis, prognosis, and treatment of addiction. TMH: None. JEK: JEK is a paid consultant for Merck as member of a data monitoring committee. EOJ: None. DBH: None.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carnes, M.U., Quach, B.C., Zhou, L. et al. Smoking-informed methylation and expression QTLs in human brain and colocalization with smoking-associated genetic loci. Neuropsychopharmacol. (2024). https://doi.org/10.1038/s41386-024-01885-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41386-024-01885-4
- Springer Nature Switzerland AG