
Abstract
Neutrophils are emerging as an important player in skeletal muscle injury and repair. Neutrophils accumulate in injured tissue, thus releasing inflammatory factors, proteases and neutrophil extracellular traps (NETs) to clear muscle debris and pathogens when skeletal muscle is damaged. During the process of muscle repair, neutrophils can promote self-renewal and angiogenesis in satellite cells. When neutrophils are abnormally overactivated, neutrophils cause collagen deposition, functional impairment of satellite cells, and damage to the skeletal muscle vascular endothelium. Heterotopic ossification (HO) refers to abnormal bone formation in soft tissue. Skeletal muscle injury is one of the main causes of traumatic HO (tHO). Neutrophils play a pivotal role in activating BMPs and TGF-β signals, thus promoting the differentiation of mesenchymal stem cells and progenitor cells into osteoblasts or osteoclasts to facilitate HO. Furthermore, NETs are specifically localized at the site of HO, thereby accelerating the formation of HO. Additionally, the overactivation of neutrophils contributes to the disruption of immune homeostasis to trigger HO. An understanding of the diverse roles of neutrophils will not only provide more information on the pathogenesis of skeletal muscle injury for repair and HO but also provides a foundation for the development of more efficacious treatment modalities for HO.
Similar content being viewed by others


Introduction
Skeletal muscle, which is also known as striated muscle, is the most abundant tissue in the human body1,2. Skeletal muscle injury is frequently clinically caused by trauma, ischemia-reperfusion, burns, and even physical exercise but has not been linked to a high-efficiency treatment until now2. Neutrophils are the first infiltrating immune cells in damaged tissue and play diverse roles in the inflammatory response3. Neutrophils are the first type of cell to defend against invading infections caused by microorganisms; moreover, they are also essential for regulating the process of tissue repair and regeneration4. Over the past few years, an increasing number of studies have focused on the complex crosstalk between skeletal muscle and neutrophils in acute injuries and chronic diseases1. Infiltrated neutrophils exacerbate the original damage by assisting in the removal of damaged tissue and the release of free radicals and protein hydrolases1. Thus, overactive neutrophils can be significantly destructive and can lead to severe tissue injury; moreover, they can exacerbate skeletal muscle damage. Interestingly, neutrophils have also been shown to contribute to tissue repair in different organs, including the liver, lung, and bone5. Different subtypes of neutrophils may have different functions during tissue repair, and more efficient phagocytosis or cytokine production and dysregulation of neutrophil heterogeneity may lead to impaired wound healing6.
Heterotopic ossification (HO) is defined as the formation of bone in soft tissues and joints7. HOs are broadly divided into traumatic HO (tHO), neurogenic HO (NHO), and genetic HO. It is commonly recognized as being a complication after trauma, surgery, spinal cord injury (SCI), or other injuries. Patients with HO suffer from joint ankylosis, which is difficult to treat, and muscle pain. tHO is most commonly induced by tissue injury, followed by inflammation and subsequent events, which inappropriately activate osteogenic or osteochondrogenic mechanisms8. Inflammatory factors recruit immune cells to create a microenvironment that serves as a vital “niche” in HO initiation and progression. A heterogeneous population of cells responds to immune cells recruited by inflammatory osteoplastic signals to initiate the bone formation process9. Muscle injury and its associated inflammation appear to be indispensable triggers for HO10; however, the injury-induced osteogenic mechanism has not yet been identified. The role of macrophages in the HO process has been explored in detail9. However, as neutrophils are the earliest recruited immune cells in injured tissues, the role of neutrophils in the HO-associated inflammatory process remains unclear, and neutrophils are considered to be able to recruit macrophages. Neutrophils and neutrophil extracellular traps (NETs) reportedly promote osteogenesis in skeletal muscle injury and play a role in the HO process11. Moreover, there is a crosstalk between neutrophils and macrophages, as well as nerve signaling, which represents the major drivers of HO.
Herein, we review the molecular roles of neutrophils in skeletal muscle injury, repair, and the process of HO. The elucidation of the vital role of neutrophils provides a theoretical basis for treatment by targeting neutrophils in skeletal muscle injury-related diseases and HO.
Neutrophils in skeletal muscle injury, repair and regeneration
Neutrophils, which originate from the bone marrow, constitute approximately 60–70% of the total leukocyte population3,12. In the classical view, neutrophils are generated from hematopoietic stem cells and differentiate into subpopulations of neutrophil killers and neutrophil capsers13,14. Granulocyte colony-stimulating factor (G-CSF) is a critical regulator of neutrophil expansion and mobilization from the bone marrow15. Furthermore, neutrophil recruitment to sites of inflammation also affects the overall frequency of neutrophils16. When they are generated in the bone marrow, neutrophils survive for only 6–8 h in circulation, after which they are cleared13,17. Neutrophils play one of the most well-known roles in inflammatory and immune processes and are also extremely diverse. Neutrophils are effective antimicrobial cells and represent the first line of cellular defense against infection by deploying antimicrobial elements, thus resulting in tissue damage12. After being recognized, neutrophils activate multiple antimicrobial mechanisms, including phagocytosis of cellular debris; degranulation of antimicrobial enzymes, such as neutrophil elastase (NE) and myeloperoxidase (MPO); DNA webs; and NETs1,17. The role of neutrophils in tissue injury and regeneration, in addition to infectious diseases, is becoming better understood18,19,20. For example, neutrophils can significantly enhance angiogenesis to promote the healing of fractures21. In conclusion, during their short life, the phenotype of neutrophils substantially changes with changes in the environment.
Skeletal muscle is physiologically important to humans and suffers more damage than other human tissues under normal conditions22. Skeletal muscle injuries can be divided into acute and chronic injuries. Acute injuries are caused by perturbations applied over a short period of time, such as lacerations, contusions, frigidity, burns, or exposure to toxins. In injured skeletal muscle, pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) signals are released, and various immune cells are recruited23. After skeletal muscle injury, the activated complement system and leaky muscle cells recruit neutrophils to participate in anti-infection reactions17,24. Neutrophils first remove tissue fragments and microorganisms from necrotic and atrophic skeletal muscle, which is beneficial for subsequent skeletal muscle repair. Moreover, neutrophil-derived cytokines, proteolytic enzymes, and NETs are also involved in this process25.
The repair of skeletal muscle involves inflammatory responses, muscle fiber regeneration, angiogenesis, and extracellular matrix (ECM) remodeling26. Infiltrated neutrophils release vascular endothelial growth factor (VEGF), and matrix metalloproteinase-9 (MMP-9) after the MAPK signaling pathway is activated25,27,28,29, thus promoting angiogenesis in injured muscle tissue. Moreover, neutrophils produce secretory leukocyte protease inhibitors (SLPIs) to accelerate wound healing after the NF-κB signaling pathway is activated30. Satellite cells mediate the initiation of muscle regeneration, regenerate damaged muscle fibers, and restore muscle contractility and metabolism26. In response to trauma, satellite cells are activated to enter the cell cycle, proliferate briefly, and further differentiate into new muscle tubes or fuse with damaged muscle fibers to repair muscle injury. The infiltration of neutrophils is also beneficial for muscle regeneration through the activation of satellite cells via the STAT pathway31,32,33,34. At the later stage of skeletal muscle repair, neutrophils gradually undergo apoptosis or migration, and apoptotic neutrophils are engulfed by macrophages, which initiates a feedback repair program and releases the tissue repair factors TGF-β and IL-10 through the TGF-β/Smad2 signaling pathway and AKT/GSK3β pathway, respectively. This subsequently promotes the remodeling of the ECM of skeletal muscle and is beneficial for restoring the normal contraction and relaxation function of skeletal muscle35 (Fig. 1).

Neutrophils are mobilized by complement to migrate toward injured skeletal muscle tissue to eliminate LPS and muscle debris at the injury site. Following their recruitment, neutrophils release TNF-α, IFN-γ, and NETs to affect damaged skeletal muscle. In the repair phase, neutrophils can enhance the renewal and proliferation of satellite cells through the STAT signaling pathway, thereby contributing to the regeneration of damaged muscle tissue. Moreover, neutrophils release VEGF, MMP-9, and IL-37 to promote angiogenesis to provide nutrients that are necessary for tissue regeneration. After neutrophil apoptosis, macrophages engulf apoptotic remnants, thus triggering the secretion of TGF-β and IL-10, which act on skeletal muscle to restore normal contraction and relaxation.
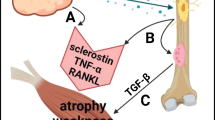
However, the overactivation of neutrophils, which results in persistent inflammatory activation, leads to tissue damage. Continuous inflammatory reactions inhibit the repair of injured skeletal muscle and cause muscle atrophy2. Afterward, the overreaction affects the release of superoxide anion radicals and/or hydrogen peroxide by neutrophils, which leads to further tissue damage36. After skeletal muscle atrophy or necrosis after injury, due to the deposition of fibrinogen, vascular permeability in diseased skeletal muscle increases, which accelerates the process of skeletal muscle repair33. However, fibrinogen also promotes the recruitment of neutrophils through interactions with the macrophage-1-antigen integrin receptor, which may lead to further overactivation of neutrophils and aggravate skeletal muscle injury37. Reactive oxygen species (ROS) released by neutrophils through the NF-κB signaling pathway are beneficial for muscle fiber degeneration and vascular changes caused by ischemia-reperfusion injury36,38. However, excessive ROS damages the vascular endothelium in muscle tissue to hinder the regeneration process of muscle39,40. Overactive neutrophils secrete IL-17 and TGF-β, which leads to the activation of stellate cells and the proliferation of adipose and connective tissue, thus ultimately limiting the function of repaired skeletal muscle41,42,43,44,45. The persistent infiltration of neutrophils in degenerative volumetric muscle loss injuries has been shown to contribute to reducing the number of muscle satellite cells that are needed for muscle cell regeneration, which was also attenuated by the JAK/STAK signaling pathway46. NETs also play a very special role in the damage of neutrophils to skeletal muscle. It is generally believed that NETs can reverse inflammation by degrading cytokines and chemokines and hinder the excessive recruitment of inflammatory cells47. However, excessive NETs can lead to tissue injury. In the presence of NETs, MMPs are released through the MAPK signaling pathway, thus leading to skeletal muscle reinjury48. MPO and NE promote the prolongation of inflammatory reactions, lead to oxidative stress, and delay the repair of skeletal muscle injury17 (Fig. 2).

Following skeletal muscle injury, neutrophils release ROS, NETs, TGF-β, IL-17, and proteases through complement recruitment to the injured site. However, the overactivation of neutrophils can result in continuous infiltration of neutrophils and the formation of NETs at the injured site. Collagen deposition and impaired regeneration of satellite cells occur through the release of IL-17, TGF-β, MPO, NE, and JAK/STAT signals. Furthermore, the excessive release of ROS and activation of NF-κB in neutrophils can directly damage the vascular endothelium in muscle tissue, thus hindering muscle repair and regeneration and eventually causing muscle atrophy.
Skeletal muscle injury to HO
HO requires inducible bone stem or progenitor cells and an ectopic environment conducive to osteogenesis49. Burns and mechanical injuries are major contributors to trauma-induced skeletal muscle damage and may further cause tHO50. During skeletal muscle injury, the first recruited immune cells are T-helper cells and neutrophils, after which macrophages dominate infiltration after approximately 24 h51,52. Neutrophils secrete TNF-α, IL-1β, and IL-1α, thus creating an inflammatory microenvironment53,54,55,56. This represents the initial phase of muscle repair, followed by two subsequent phases of muscle regeneration and fibrosis, with muscle repair being a delicate equilibrium between muscle regeneration and fibrosis. Nevertheless, the overactivation of neutrophils may heighten the probability of scarring or fibrosis by aggravating muscle damage, which is the primary factor contributing to the gradual decline in muscle functionality and the occurrence of HO57,58,59. Thus, the appropriate presence and activation of neutrophils are crucial for the management of muscle injury and HO.
Neutrophil-derived TGF-β and BMP signaling drive osteogenic differentiation. TGF-β signaling regulates scleraxis expression in skeletal muscle fibroblasts, thereby facilitating proliferation and collagen type-I synthesis, both of which are critical for effective tissue repair60. Furthermore, BMP signaling leads to the differentiation of MSCs toward the osteogenic lineage61. This mechanism is a potential method of triggering tHO in vivo, particularly when neutrophils are overactivated and release BMP in conjunction with TGF-β. Furthermore, TGF-β can bind to Smad2/3, which is activated to regulate inflammation when ActA is combined with BMP receptors, thus suggesting that BMP associates with TGF-β to induce strong tHO62,63. Smad1/5/8 can be phosphorylated and translocated to the nucleus in the presence of BMP-2 signaling alone in C2C12 cells, where it converts C2C12 cells to osteoblasts61. Interestingly, TGF-β inhibits HO by reducing the nuclear translocation of the downstream protein Smad1/5/8 and preventing activation of the Wnt pathway64. Janna et al. used metabolic profiling to demonstrate that itaconate, which is produced by mature neutrophils at injury sites, is a metabolite that is differentially abundant between HO and non-HO in burn/tenotomy models of the Achilles tendon65. These mature neutrophils contribute to prolonged inflammation by secreting the cytokines CCL2 and IL-1β. Moreover, itaconate-producing neutrophils return to the bone marrow for degradation by macrophages and turnover of hematopoiesis to the myeloid lineage.
Trauma can also lead to a local and systemic inflammatory state with elevated inflammatory cytokines, such as TNFα, IL-1β, and IL-666. The persistence of the inflammatory microenvironment is a significant alteration in the development of HO, thus allowing for neutrophils and macrophages to be overactivated and further leading to abnormal osteogenic differentiation of MSCs by affecting the balance of bone formation and resorption through the NF-κB signaling pathway, thereby promoting HO66,67.
Overactivation of neutrophils causes a traumatic site to develop into a hypoxic environment68,69. Additionally, neutrophils upregulate the transcription factor SOX-9, thereby promoting chondrocyte differentiation70,71. Consequently, chondrocytes undergo excessive proliferation and initiate the formation of a cartilage matrix, thus ultimately leading to the occurrence of HO72,73,74. Furthermore, hypoxia-inducible factor (HIF) serves as a regulator of BMP-2-induced endochondral osteogenesis in fetal limb culture75. Significantly, the synergistic relationship between HIF and VEGF amplifies the expression of endothelial osteogenic growth factors, thereby facilitating osteoblast differentiation but also attracting bone-forming stem cells to the injured tissue72 (Fig. 3).

Following skeletal muscle injury, the infiltration of neutrophils and macrophages results in a localized inflammatory and hypoxic microenvironment. These neutrophils and macrophages consistently release cytokines, including TNF-α, TGF-β1, and BMP, which facilitate the differentiation of MSCs and OPCs (markers) into osteoblasts and chondrocytes by upregulating SOX-9, SMAD2/3, Runx2, and other factors. This process culminates in the development of ectopic ossification.
As shown above, during the repair process of trauma-induced skeletal muscle injury, complex interactions among inflammatory cells, neutrophils, macrophages, and a hypoxic environment are the main causes of tHO. Importantly, compared with those in the blood circulation or bone marrow, neutrophils at the site of HO strongly express inflammatory cytokines to prolong local inflammation and support abnormal cell differentiation. This finding implies that there are local factors at the injury site that mediate the differences in neutrophils. Moreover, macrophages are extremely versatile cells, and the function of these cells mainly depends on the local environment. HO site neutrophils homed to the bone marrow and promoted the myeloid differentiation of bone marrow stem cells. Locally present neutrophils alter the phenotype of subsequently recruited macrophages, thus making them a major contributor to subsequent HO progression. Although macrophages and neutrophils are capable of producing cytokines, the mechanism of how they are abnormally activated by trauma and how they produce sufficient amounts of cytokines (such as BMP and TGF-β) to cause tHO in vivo remain open questions.
Molecular mechanism of neutrophils in tHO
tHO requires three osteogenic conditions: osteogenic signal induction factors, osteogenic precursor cells, and an appropriate local microenvironment76,77. BMP signaling is one of the most common osteogenic signaling pathways for HO-forming progenitor cells78. Neutrophils significantly regulate major osteogenic markers, such as BMP 2-5, TGF-β2, RUNX2, and extracellular matrix (ECM) proteins79. Moreover, neutrophils activate MSCs and cause osteogenic differentiation accompanied by increased levels of IL-1α and TGF-β80. The neutrophil-mediated inflammatory response enhances the chondrocyte differentiation of MSCs84.
Neutrophils are also able to influence osteoblasts and osteoclasts through Il-1β, Il-6, IFN-γ, and TNF-α90. Furthermore, the presence of neutrophils eliminates pathogenic microorganisms in a timely manner, thus potentially creating conditions conducive to HO. Davisbk et al. demonstrated that macrophages stimulated by LPS are more prone to convert to M1 macrophages through the IRF/STAT signaling pathway, which has the potential to become osteoclasts91. However, overactivated neutrophils convert macrophages to M2 macrophages, which triggers osteogenesis via MSCs82,91.
The mechanisms by which neutrophils interact with other cells in the microenvironment, such as osteoblasts and osteoclasts, are necessary to elicit HO and still require further investigation. Additionally, when considering the limited lifespan of neutrophils, the development of in vitro models and enhanced culture methods for studying neutrophils may contribute to a more comprehensive understanding of their involvement in the formation of HO (Fig. 4).

The overactivation of neutrophils can contribute to the occurrence of HO. Neutrophils interact with MSCs via an IL-Lα, IL-8, and TNF-α loop and release SDF-1α to stimulate the chondrocyte differentiation of MSCs through the CXCR4-PI3K/Akt/β-catenin pathway. Overactivation of neutrophils also leads to significant local infiltration of NETs, which promotes TLR9 signaling and directly facilitates the formation of HO. Furthermore, neutrophils directly eliminate LPS, thereby creating an environment conducive to the shift of macrophages from the M1 to the M2 osteogenic phenotype, which subsequently enhances the development of HO.
Targeting neutrophils in skeletal muscle injury and HO therapies
Due to the critical and complex roles of neutrophils in skeletal muscle injury and repair in HO, we need to distinguish between secondary responses and potentially deleterious side effects. A better understanding of the roles of neutrophils may help us to consider them as promising therapeutic targets (Fig. 5).

The treatment of skeletal muscle injury and HO is achieved by targeting neutrophils and the resulting NETs in the injured microenvironment. Through targeted reduction of neutrophil chemokines, the transplantation of umbilical cord MSCs (UMSCs) and mechanical loading can reduce the number of damaged neutrophils to reduce muscle injury. Moreover, by reducing the adhesion of neutrophils, a hydrogel known as CIC can be used to control injury-induced inflammation. In addition, batroxobin, hydroxychloroquine (HCQ), and the TLR9 inhibitor OPN-2088 can inhibit HO by clearing NETs.
Cyclic compressive loading within a specific range of forces directs the rapid clearance of neutrophils to improve muscle regeneration92. Another promising route for combating the inflammatory response caused by muscle injury is to transplant umbilical cord MSCs (UMSCs). Studies have shown that UMSCs can attenuate neutrophil-derived acute inflammation by ameliorating early-onset neutrophil infiltration and activation, thereby mitigating the extent of inflammation53. Some drugs have also been found to be effective in the treatment of muscle injury. A hydrogel system consisting of CD146, IGF-1, collagen I/III, and poloxamer 407 (termed CIC gel), which secretes CD146 to effectively promote efferocytosis and neutrophil adhesion, could be a promising tool for impeding muscle inflammation93. Similarly, a neutrophil-mediated delivery system loaded with liposomes was discovered to have the potential to deliver anti-inflammatory drugs such94 as methotrexate (a potent immunosuppressive agent used to treat inflammation) without impairing neutrophil viability and chemotaxis, through which neutrophils can retain their ability to rapidly migrate to inflamed tissue and subsequently release drug-loaded liposomes, thus causing the intended anti-inflammatory effect94. Nunez demonstrated that reducing the overall neutrophil abundance at the injury site also mitigated HO formation, whether through pharmacological treatment with hydroxychloroquine (HCQ), treatment with the TLR9 inhibitor OPN-2088, or mechanical treatment with limb unloading11. Several studies have demonstrated the ability of batroxobin to abrogate neutrophil extracellular traps, which may be used to suppress inflammation and expedite myoblast regeneration95. However, the mechanism by which neutrophils function in skeletal muscle injury leading to HO remains largely unknown, and more research is needed to fill in the knowledge gaps for neutrophils to be viable targets.
Conclusion and future perspective
Neutrophils are the initial immune cells recruited at the site of skeletal muscle injury. They eliminate skeletal muscle debris and microorganisms through the release of inflammatory factors, protein hydrolases, and NETs. Following anti-infection measures, NETs facilitate the resolution of inflammation by degrading cytokines and chemokines, thus impeding the excessive recruitment of inflammatory cells. Neutrophils can generate VEGF and MMP-9 to promote tissue repair. Additionally, apoptotic neutrophils can stimulate macrophages, thus leading to the release of TGF-β and IL-10, thereby facilitating the remodeling of the extracellular matrix in skeletal muscle and aiding in the restoration of normal contractile and diastolic functions. However, neutrophil overactivation and the persistence of NETs can result in prolonged inflammation, which ultimately impairs the functionality of satellite cells and limits the overall repair process. Skeletal muscle injury is a prominent contributor to tHO. Neutrophil overactivation and the resulting immunological disturbances associated with skeletal muscle injury can significantly stimulate the production of NETs, as well as the activation of the TGF-β1 and BMP signaling pathways in local tissue. This dysfunctional inflammatory microenvironment serves as a catalyst for the osteogenic differentiation of various stem cells, including MSCs. Neutrophils can exert systemic effects on osteoblasts and osteoclasts through a systemic inflammatory response. Moreover, neutrophils also contribute to HO progression by affecting NGF signaling and myeloid hematopoietic bias. However, it is important to note that neutrophils play dual roles in the early stages of muscle injury, either by promoting muscle repair or contributing to the development of HO. In view of the great heterogeneity of circulating neutrophils and neutrophils at the site of HO, the local upstream signaling mediating this heterogeneity is worthy of investigation. In conclusion, neutrophils themselves may play a considerable role in the initiation and progression of HO, and it is likely that they are also capable of regulating the role of other cells and neural signaling. To date, studies of neutrophils in HO are still scarce, and the mechanisms underlying their interaction with other cells and secreted cytokines still need to be elucidated. For example, the mechanisms by which neutrophils are overactivated in the injured microenvironment and how NETs are specifically localized at the injury site remain unknown and need to be discussed in the future. This cell interaction may occur as early as in the bone marrow and is of great concern. In summary, neutrophils potentially exert a significant influence on the onset and progression of HO, and they may also possess the ability to modulate the functions of other immune cells. Currently, investigations pertaining to neutrophils in the context of HO are limited, and further elucidation is required regarding their interactions with other cells and the cytokines that they secrete, as well as for expanding the study to the whole body rather than just the injured site. Moreover, given the transient lifespan of neutrophils, enhancing in vitro models and culture techniques for these cells would positively contribute to a more comprehensive understanding of their role in HO.
References
You, Z. et al. Molecular feature of neutrophils in immune microenvironment of muscle atrophy. J. Cell. Mol. Med. 26, 4658–4665 (2022).
Howard, E. E., Pasiakos, S. M., Blesso, C. N., Fussell, M. A. & Rodriguez, N. R. Divergent roles of inflammation in skeletal muscle recovery from injury. Front. Physiol. 11, 87 (2020).
Nemeth, T., Sperandio, M. & Mocsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 19, 253–275 (2020).
Bouchery, T. & Harris, N. Neutrophil-macrophage cooperation and its impact on tissue repair. Immunol. Cell Biol. 97, 289–298 (2019).
Peiseler, M. & Kubes, P. More friend than foe: the emerging role of neutrophils in tissue repair. J. Clin. Investig. 129, 2629–2639 (2019).
Liu, Y. et al. Neutrophil heterogeneity and aging: implications for COVID-19 and wound healing. Front. Immunol. 14, 1201651 (2023).
Hwang, C. D. et al. Contemporary perspectives on heterotopic ossification. JCI Insight 7, e158996 (2022).
Meyers, C. et al. Heterotopic ossification: a comprehensive review. JBMR 3, e10172 (2019).
Huang, Y. et al. Macrophages in heterotopic ossification: from mechanisms to therapy. NPJ Regen. Med. 6, 70 (2021).
Li, L. & Tuan, R. S. Mechanism of traumatic heterotopic ossification: In search of injury-induced osteogenic factors. J. Cell. Mol. Med. 24, 11046–11055 (2020).
Nunez, J. H. et al. Neutrophil and NETosis modulation in traumatic heterotopic ossification. Ann. Surg. 278, e1289–e1298 (2023).
Burn, G. L., Foti, A., Marsman, G., Patel, D. F. & Zychlinsky, A. The neutrophil. Immunity 54, 1377–1391 (2021).
Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182, (2006).
Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 371, 531–539 (2018).
Manz, M. G. & Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 14, 302–314, (2014).
Mayadas, T. N., Cullere, X. & Lowell, C. A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218 (2014).
Tulangekar, A. & Sztal, T. E. Inflammation in Duchenne muscular dystrophy-exploring the role of neutrophils in muscle damage and regeneration. Biomedicines 9, 1366 (2021).
Van Avondt, K. et al. Neutrophils in aging and aging-related pathologies. Immunol. Rev. 314, 357–375 (2022).
Shirakawa, K. & Sano, M. Neutrophils and neutrophil extracellular traps in cardiovascular disease: an overview and potential therapeutic approaches. Biomedicines 10, 1850 (2022).
Nolan, E. & Malanchi, I. Connecting the dots: neutrophils at the interface of tissue regeneration and cancer. Semin. Immunol. 57, 101598 (2021).
Ishida, K. et al. Bone regeneration properties of granulocyte colony-stimulating factor via neovascularization and osteogenesis. Tissue Eng. Part A 16, 3271–3284 (2010).
Orchard, J. & Seward, H. Epidemiology of injuries in the Australian Football League, seasons 1997–2000. Br. J. Sports Med. 36, 39–44 (2002).
Pittman, K. & Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 5, 315–323, (2013).
Dufaux, B. & Order, U. Complement activation after prolonged exercise. Clin. Chim. Acta 179, 45–49 (1989).
Jones, H. R., Robb, C. T., Perretti, M. & Rossi, A. G. The role of neutrophils in inflammation resolution. Semin. Immunol. 28, 137–145, (2016).
Yang, W. & Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 13, 25–32 (2018).
Dalli, J. et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol. Cell. Proteom. 12, 2205–2219 (2013).
Gong, Y. & Koh, D. R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 339, 437–448 (2010).
Ardi, V. C., Kupriyanova, T. A., Deryugina, E. I. & Quigley, J. P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl Acad. Sci. USA 104, 20262–20267, (2007).
Ghasemlou, N. et al. Beneficial effects of secretory leukocyte protease inhibitor after spinal cord injury. Brain 133, 126–138 (2010).
Pizza, F. X., Peterson, J. M., Baas, J. H. & Koh, T. J. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 562, 899–913 (2005).
Sugimoto, M. A., Sousa, L. P., Pinho, V., Perretti, M. & Teixeira, M. M. Resolution of inflammation: what controls its onset? Front. Immunol. 7, 160 (2016).
Dort, J., Fabre, P., Molina, T. & Dumont, N. A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 4761427 (2019).
Ratnayake, D. et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nature 591, 281–287 (2021).
Philippou, A., Maridaki, M., Theos, A. & Koutsilieris, M. Cytokines in muscle damage. Adv. Clin. Chem. 58, 49–87 (2012).
Romson, J. L. et al. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 67, 1016–1023 (1983).
Tulangekar, A. & Sztal, T. E. J. B. Inflammation in Duchenne muscular dystrophy—exploring the role of neutrophils in muscle damage and regeneration. Biomedicines 9, 1366 (2021).
Korthuis, R. J., Grisham, M. B. & Granger, D. N. Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am. J. Physiol. 254, H823–H827 (1988).
Chen, H., Song, Y. S. & Chan, P. H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow. Metab. 29, 1262–1272 (2009).
Terrill, J. R. et al. Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy. Redox Biol. 9, 276–286 (2016).
Puttur, F., Gregory, L. G. & Lloyd, C. M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 97, 246–257 (2019).
Orchard, J. & Best, T. M. The management of muscle strain injuries: an early return versus the risk of recurrence. Clin. J. Sport Med. 12, 3–5 (2002).
Tidball, J. G., Welc, S. S. & Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 8, 1313–1356 (2018).
Carpenter, S. & Karpati, G. Duchenne muscular dystrophy: plasma membrane loss initiates muscle cell necrosis unless it is repaired. Brain 102, 147–161, (1979).
Juban, G. et al. AMPK activation regulates LTBP4-dependent TGF-β1 secretion by Pro-inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep. 25, 2163–2176.e2166 (2018).
Larouche, J. A. et al. Neutrophil and natural killer cell imbalances prevent muscle stem cell-mediated regeneration following murine volumetric muscle loss. Proc. Natl Acad. Sci. USA 119, e2111445119 (2022).
Schauer, C. et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 20, 511–517 (2014).
Wilgus, T. A., Roy, S. & McDaniel, J. C. Neutrophils and wound repair: positive actions and negative reactions. Adv. Wound Care (N. Rochelle) 2, 379–388 (2013).
Kaplan, F. S., Glaser, D. L., Hebela, N. & Shore, E. M. Heterotopic ossification. J. Am. Acad. Orthop. Surg. 12, 116–125 (2004).
Smith, C., Kruger, M. J., Smith, R. M. & Myburgh, K. H. The inflammatory response to skeletal muscle injury. Sports Med. 38, 947–969 (2008).
Loell, I. & Lundberg, I. E. Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J. Intern. Med. 269, 243–257 (2011).
McClung, J. M., Davis, J. M. & Carson, J. A. Ovarian hormone status and skeletal muscle inflammation during recovery from disuse in rats. Exp. Physiol. 92, 219–232 (2007).
Su, W. H. et al. Human umbilical cord mesenchymal stem cells extricate bupivacaine-impaired skeletal muscle function via mitigating neutrophil-mediated acute inflammation and protecting against fibrosis. Int. J. Mol. Sci. 20, 4312 (2019).
Tidball, J. G. Inflammatory cell response to acute muscle injury. Med. Sci. Sports Exerc. 27, 1022–1032 (1995).
Fielding, R. A. et al. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. 265, R166–172, (1993).
Lämmermann, T. et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498, 371–375 (2013).
Haffner-Luntzer, M., Fischer, V., Prystaz, K., Liedert, A. & Ignatius, A. The inflammatory phase of fracture healing is influenced by estrogen status in mice. Eur. J. Med. Res. 22, 23 (2017).
Kovtun, A. et al. The crucial role of neutrophil granulocytes in bone fracture healing. Eur. Cells Mater. 32, 152–162 (2016).
Recknagel, S. et al. Systemic inflammation induced by a thoracic trauma alters the cellular composition of the early fracture callus. J. Trauma Acute Care Surg. 74, 531–537 (2013).
Davies, O. G. et al. Defining the balance between regeneration and pathological ossification in skeletal muscle following traumatic injury. Front. Physiol. 8, 194 (2017).
Xu, Y. et al. Heterotopic ossification: clinical features, basic studies, and mechanical stimulations. Front. Cell Dev. Biol. 10, 770931 (2022).
Zhang, F. et al. PPARγ and MyoD are differentially regulated by myostatin in adipose-derived stem cells and muscle satellite cells. Biochem. Biophys. Res. Commun. 458, 375–380 (2015).
Bernabeu, C., Lopez-Novoa, J. M. & Quintanilla, M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 1792, 954–973 (2009).
Guerrero, F. et al. TGF-β prevents phosphate-induced osteogenesis through inhibition of BMP and Wnt/β-catenin pathways. PLoS ONE 9, e89179 (2014).
Crossley, J. L. et al. Itaconate-producing neutrophils regulate local and systemic inflammation following trauma. JCI Insight 8, e169208 (2023).
Sung Hsieh, H. H. et al. Evaluation of Salivary cytokines for diagnosis of both trauma-induced and genetic heterotopic ossification. Front. Endocrinol. (Lausanne) 8, 74 (2017).
Convente, M. R. et al. Depletion of mast cells and macrophages impairs heterotopic ossification in an Acvr1(R206H) mouse model of fibrodysplasia ossificans progressiva. J. Bone Min. Res. 33, 269–282 (2018).
Winning, S. & Fandrey, J. Oxygen sensing in innate immune cells: how inflammation broadens classical hypoxia-inducible factor regulation in myeloid cells. Antioxid. Redox Signal. 37, 956–971 (2022).
Lim, C. S., Kiriakidis, S., Sandison, A., Paleolog, E. M. & Davies, A. H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 58, 219–230 (2013).
Winkler, S. et al. The impact of hypoxia on mesenchymal progenitor cells of human skeletal tissue in the pathogenesis of heterotopic ossification. Int. Orthop. 39, 2495–2501 (2015).
Robins, J. C. et al. Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone 37, 313–322 (2005).
Stegen, S., van Gastel, N. & Carmeliet, G. Bringing new life to damaged bone: the importance of angiogenesis in bone repair and regeneration. Bone 70, 19–27 (2015).
Qureshi, A. T. et al. Early characterization of blast-related heterotopic ossification in a rat model. Clin. Orthop. Relat. Res. 473, 2831–2839 (2015).
Agarwal, S. et al. Diminished chondrogenesis and enhanced osteoclastogenesis in leptin-deficient diabetic mice (ob/ob) impair pathologic, trauma-induced heterotopic ossification. Stem Cells Dev. 24, 2864–2872 (2015).
Zhou, N. et al. HIF-1α as a regulator of BMP2-induced chondrogenic differentiation, osteogenic differentiation, and endochondral ossification in stem cells. Cell. Physiol. Biochem. 36, 44–60 (2015).
Chalmers, J., Gray, D. H. & Rush, J. Observations on the induction of bone in soft tissues. J. Bone Jt. Surg. Br. 57, 36–45 (1975).
Yin, N. et al. MiR-135-5p promotes osteoblast differentiation by targeting HIF1AN in MC3T3-E1 cells. Cell. Mol. Biol. Lett. 24, 51 (2019).
Shore, E. M. & Kaplan, F. S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 6, 518–527, (2010).
Herath, T. D. K., Larbi, A., Teoh, S. H., Kirkpatrick, C. J. & Goh, B. T. Neutrophil-mediated enhancement of angiogenesis and osteogenesis in a novel triple cell coculture model with endothelial cells and osteoblasts. J. Tissue Eng. Regen. Med. 12, e1221–e1236 (2018).
Al-Hakami, A. et al. Cytokine physiognomies of MSCs from varied sources confirm the regenerative commitment postcoculture with activated neutrophils. J. Cell Physiol. 235, 8691–8701 (2020).
Chung, R., Cool, J. C., Scherer, M. A., Foster, B. K. & **an, C. J. Roles of neutrophil-mediated inflammatory response in the bony repair of injured growth plate cartilage in young rats. J. Leukoc. Biol. 80, 1272–1280 (2006).
Cai, B. et al. N2-Polarized neutrophils guide bone mesenchymal stem cell recruitment and initiate bone regeneration: a missing piece of the bone regeneration puzzle. Adv. Sci. (Weinh., Baden.-Wurtt., Ger.) 8, e2100584 (2021).
Qin, Q. et al. Neurovascular coupling in bone regeneration. Exp. Mol. Med. 54, 1844–1849 (2022).
Lee, S. et al. NGF-TrkA signaling dictates neural ingrowth and aberrant osteochondral differentiation after soft tissue trauma. Nat. Commun. 12, 4939 (2021).
Cherief, M. et al. TrkA+ neurons induce pathologic regeneration after soft tissue trauma. Stem Cells Transl. Med. 11, 1165–1176 (2022).
Cherief, M. et al. TrkA-mediated sensory innervation of injured mouse tendon supports tendon sheath progenitor cell expansion and tendon repair. Sci. Transl. Med. 15, eade4619 (2023).
Xu, J. et al. NGF-p75 signaling coordinates skeletal cell migration during bone repair. Sci. Adv. 8, eabl5716 (2022).
Sas, A. R. et al. A new neutrophil subset promotes CNS neuron survival and axon regeneration. Nat. Immunol. 21, 1496–1505 (2020).
Jerome, A. D. et al. Characterization of Zymosan-modulated neutrophils with neuroregenerative properties. Front. Immunol. 13, 912193 (2022).
Geissler, S. et al. Loss of murine Gfi1 causes neutropenia and induces osteoporosis depending on the pathogen load and systemic inflammation. PLoS ONE 13, e0198510 (2018).
Olmsted-Davis, E., Mejia, J., Salisbury, E., Gugala, Z. & Davis, A. R. A population of M2 macrophages associated with bone formation. Front. Immunol. 12, 686769 (2021).
Seo, B. R. et al. Skeletal muscle regeneration with robotic actuation-mediated clearance of neutrophils. Sci. Transl. Med. 13, eabe8868 (2021).
Lee, W. et al. Thermosensitive hydrogel harboring CD146/IGF-1 nanoparticles for skeletal-muscle regeneration. ACS Appl. Bio Mater. 4, 7070–7080 (2021).
Che, J. et al. Neutrophils enable local and non-invasive liposome delivery to inflamed skeletal muscle and ischemic heart. Adv. Mater. 32, e2003598 (2020).
Masuda, H. et al. Batroxobin accelerated tissue repair via neutrophil extracellular trap regulation and defibrinogenation in a murine ischemic hindlimb model. PLoS ONE 14, e0220898 (2019).
Acknowledgements
The figures were created with BioRender.com. This work was supported by the National Nature Science Foundation of China (31900852 to H.L.) and the Nature Science Foundation of Jiangxi Province of China (20224ACB206024, 20232BAB206081 and 20232BCJ23008 to H.L.).
Author information
Authors and Affiliations
Contributions
H.L. and L.-Z.S. conceived the study. L.-Z.S. and X.-L.Z. wrote the manuscript. Y.-D.D. participated in the figure production. L.-Z.S., Y.-D.D., X.-L.Z., and H.L. improved the quality of the manuscript. All of the authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shu, LZ., Zhang, XL., Ding, YD. et al. From inflammation to bone formation: the intricate role of neutrophils in skeletal muscle injury and traumatic heterotopic ossification. Exp Mol Med (2024). https://doi.org/10.1038/s12276-024-01270-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s12276-024-01270-7
- Springer Nature Limited