

Abstract
Identification of new biomarkers and drug targets for chronic kidney disease (CKD) is required for the development of more effective therapy. Here we report an association between expression of reticulon 1 (RTN1) and severity of CKD. An isoform-specific increase in the expression of RTN1A is detected in the diseased kidneys from mice and humans, and correlates inversely with renal function in patients with diabetic nephropathy. RTN1 overexpression in renal cells induces ER stress and apoptosis, whereas RTN1 knockdown attenuates tunicamycin-induced and hyperglycaemia-induced ER stress and apoptosis. RTN1A interacts with PERK through its N-terminal and C-terminal domains, and mutation of these domains prevents this effect on ER stress. Knockdown of Rtn1a expression in vivo attenuates ER stress and renal fibrosis in mice with unilateral ureteral obstruction, and also attenuates ER stress, proteinuria, glomerular hypertrophy and mesangial expansion in diabetic mice. Together, these data indicate that RTN1A contributes to progression of kidney disease by inducing ER stress.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) affects ∼10% of US adults. The incidence and prevalence of this condition are increasing worldwide1. Therapeutic options for CKD are limited and at best only offer partial protection against disease progression2,3,4. Therefore, there is an urgent need to identify key mediators of CKD progression for the development of more effective therapy to halt its progression.
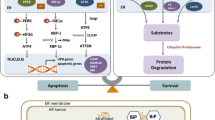
In this study, to identify such potential mediators, we profiled the renal gene expression of a murine model of HIV-associated nephropathy (HIVAN), which has progressive CKD. Genes whose level of expression correlated with severity of renal injury were identified and among them was Rtn1, which encodes for an endoplasmic reticulum (ER)-associated protein called reticulon 1 (RTN1). The reticulon family has four members: RTN1, RTN2, RTN3 and RTN4. Reticulons were first described in neuroendocrine cells and their function is implicated in neurodegenerative diseases. They localize primarily to the ER membrane as ER-sha** proteins. Reticulons are known to induce apoptosis, inhibit axon regeneration and regulate protein trafficking5,6,7,22 and are known to be involved in endocytosis10 and initiation of apoptosis through regulation of ER stress in neuronal cells11,12. However, a biologic function of RTN1A has not been well characterized. Here we demonstrated that RTN1A plays a key role in ER stress and apoptosis of kidney cells. Our results suggest that RTN1A mediates high-glucose-induced ER stress and apoptosis of kidney cells. In addition, we found that high glucose induces RTN1A expression in kidney cells through the activation of oxidative stress pathway. These data suggest that RTN1A could be important for hyperglycaemia-induced tubular injury and progression of renal injury in kidney diseases.
To understand how RTN1A contributes to ER stress, we examined the interaction of RTN1A with PERK, a key ER stress molecule leading to the activation of apoptosis pathway. Interestingly, we found that only WT-RTN1A interacted with PERK, whereas MT-RTN1A or RTN1C did not. In addition, expression of MT-RTN1A or RTN1C was unable to induce ER stress and apoptosis in kidney cells, suggesting that both N and C terminals of RTN1A are required for its interaction with PERK and for its induction of ER stress and apoptosis in kidney cells. However, our findings are not consistent with the previous reports showing that RTN1C overexpression induces ER stress and apoptosis in neuronal cells11,12. While it is plausible that different isoforms may have cell type-specific functions, detailed mechanism of this discrepancy/inconsistency requires further studies. Future studies are also required to map the potential interactive domains between RTN1A and PERK and to understand how this interaction leads to the activation of ER stress in kidney cells.
To confirm the role of RTN1A in the progression of kidney disease, we developed a mouse model with knockdown of Rtn1a expression by shRNA. We found that knockdown of Rtn1a expression before the surgery attenuated renal fibrosis and ER stress in the UUO model. We also found that the knockdown of RTN1A expression in the mice with established kidney disease also ameliorated the progression of the disease. To verify that our findings are not a result of nonspecific off-target effects of shRNA, we validated these findings in another line of Rtn1a knockdown mice. In addition, we showed that tubular cell-specific knockdown of RTN1A also attenuated kidney injury, indicating an important/a direct role of RTN1A in tubular cell injury. Finally, knockdown of Rtn1a also ameliorated albuminuria, kidney and glomerular hypertrophy, and mesangial expansion in STZ-induced diabetic mice, suggesting that RTN1A is involved in mediating these changes of early DN. Future studies are required to confirm whether lack of RTN1A also attenuates kidney injury in an animal model of progressive DN. Taken together, this is the first in vivo evidence suggesting that RTN1A contributes to the development of kidney disease and that it may be a potential drug target for therapy for CKD progression.
A large body of evidence suggests that ER stress plays a major role in the development and progression of kidney disease including DN23,24,25. High glucose concentrations induce ER stress and apoptosis of kidney cells17. Diabetic rats exhibit enhanced kidney cell apoptosis, CHOP, JNK and Caspase-12 expression26. Diabetic CHOP knockout mice seemed to be protected from DN, because they developed less proteinuria than WT control mice27. Elevated urinary protein excretion in humans is known to be associated with tubular injury and ER stress17,28,29. In humans and rats with nephrotic syndrome, ER stress markers are elevated in tubular epithelial cells30,31. In addition, renal expression of ER stress markers in patients with established DN is higher than patients with mild DN. These results suggest an activation of ER stress response in the kidney of patients with established DN17. Over the past decade, there has been considerable interest in develo** compounds that modulate ER stress response. Chemical chaperones that improve ER folding capacity, such as 4-PBA, taurine-conjugated ursodeoxycholic acid and the ER chaperone ORP150, have been shown to reduce ER stress, restore glucose tolerance and improve insulin action and sensitivity32,33,34. 4-PBA is an aromatic short chain fatty acid that has chaperone-like activities and recent work shows that PBA attenuates kidney injury and oxidative stress in rats with DN35. Our data suggest that 4-PBA abolishes RTN1A-induced ER stress and apoptosis in HK2 cells. However, the ER stress response could have both protective and deleterious features depending on whether it is the initial adaptive response or whether prolonged and chronic. An improved understanding of the molecules regulating these processes in a cell- and disease-specific manner will help identify novel therapeutic strategies targeting ER stress to prevent the progression of kidney36disease.
In conclusion, we have shown that RTN1A expression is highly upregulated in HIVAN and DN, and that RTN1A is a key molecule mediating hyperglycaemia-induced ER stress and apoptosis of renal cells, contributing to the progression of kidney disease. Importantly, the inhibition of its expression attenuates renal fibrosis and diabetic kidney injury, suggesting that it may be a novel therapeutic target for treatment of CKD including DN.
Methods
Animal studies
All animal studies were approved by the IACUC committee of Mount Sinai School of Medicine. Genomic DNA extracted from tail clip** was used. To detect the HIV-1 transgene (Tg26), we performed standard PCR procedures using the following primers: forward 5′- ACATGAGCAGTCAGTTCTGCCGCAGAC -3′, reverse 5′- CAAGGACTCTGATGCGCAGGTGTG -3′. Thermo profile for the PCR reaction was as follows: 95 °C for 5 min followed by 35 cycles at 95 °C for 30 s, 62 °C for 45 s and 72 °C for 45 s, then 72 °C for 10 min. Only male heterozygous Tg26 in the FVB/N background were used in the study, since homozygous Tg26 mice are not viable for more than few weeks postnatally. Male diabetic db/db, nondiabetic db/m and eNOS−/− mice in B6 background were obtained from Jackson Laboratory and crossed to generate db/db;eNOS−/−mice and litter-matched control mice. The kidneys were collected from these mice for histology, western blot, real-time PCR analysis and microarray studies. Kidney disease was confirmed by measurement of proteinuria, renal function and histologic analysis.
Creation of Rtn1 knockdown mouse model
We developed a DOX-inducible RNAi model20 for Rtn1. Nine shRNA guide sequences predicted to target Rtn1 for knockdown were embedded into a miR30-based expression cassette of a retroviral DOX-inducible shRNA vector. Two sequences achieved >50% knockdown compared with a control shRNA based on both real-time PCR and western blot analyses (Supplementary Figs 13 and 14). Two lines of genetically engineered mice, each corresponding to one of the two guide sequences that achieved >50% knockdown in vitro, were generated using the ColA1 Flp/FRT recombinase-mediated targeting system. Details of construct design and transgenesis have been described previously20. Briefly, the ‘Flp-In’ targeting vector, called pCol-TGM, was configured with a GFP ‘spacer’ between a tetracycline-regulated element and the miR30-based expression cassette. Genetically engineered ‘KH2’ embryonic stem cells were used for targeted insertion of pCol-TGM to generated TGM mice. To generate CAGs;Rtn1RNAi mice, TGM mice were bred with another line of genetically engineered mice that express the rtTA under the control of the CAGs promoter. CAGs;Rtn1RNAi mice derived from KH2 ES cells are on a hybrid genetic background with contributions from C57BL/6 and 129/SV strains. To generate renal tubular epithelial cell-specific Rtn1a knockdown mice, we crossed Pax8-rtTA mice with Rtn1RNAimice to have Pax8;Rtn1RNAimice. To induce Rtn1a knockdown mice were fed 625mg/kg DOX-supplemented chow (Bio-Serv, Frenchtown, NJ) for the duration specified in the Results and figure legends.
Creation of UUO model
To create the UUO model, the left ureter of mice was exposed through a mid-abdominal incision and ligated using 4-0 silk. Sham-operated mice had their ureters exposed and manipulated but not ligated. All surgeries were performed under general anaesthesia with isofluorane. Six mice in each group were selected based on previous studies without the use of randomization and blinding37.
Creation of diabetic model by STZ injection
Induction of diabetes using STZ has been described38. Briefly, 8-week-old WT and shRNA mice were given low-dose STZ (50 mg kg−1) in 50 mmol l−1 sodium citrate buffer (pH 5.4) intraperitoneally daily for 5 days. At 2 weeks post-injection, mice were deprived of food for 6 h and fasting blood glucose level from the tail vein was measured using the One Touch Blood Glucose Monitoring System. Repeat fasting blood glucose measurements were taken every 2 weeks to verify hyperglycaemia. Diabetes mellitus was defined as sustained fasting blood glucose above 250 mg dl−1 at two distinct time points. All mice were killed at 6 months of age. Six mice in each group were selected based on our previous publication without the use of randomization and blinding38.
Urine albumin and creatinine
Twenty-four-hour urine was collected from mice using metabolic cages and urine albumin was determined using a commercial assay from Bethyl Laboratory Inc. (Houston, Texas, USA). Urine creatinine levels were measured in the same samples using the QuantiChrom Creatinine Assay Kit (DICT-500) (BioAssay Systems) according to the manufacturer’s instructions. The urine albumin excretion rate was expressed as the ratio of albumin to creatinine.
Kidney histology
Kidney samples were fixed in 10% formalin, embedded in paraffin and sectioned to 4-μm thickness. Periodic acid–Schiff (PAS)-stained sections were used to assess kidney histological features. The glomerular volume and mesangial area were determined by examining PAS-stained sections using a digitizing tablet and video camera. Briefly, digitized images were scanned and profile areas were traced using ImageJ. The mean glomerular tuft volume was determined from the mean glomerular cross-sectional area by light microscopy. The glomerular cross-sectional area was calculated on the basis of the average area of 30 glomeruli in each group, and glomerular tuft volume was calculated using the following equation: GV=(β/κ) × GA3/2, where β=1.38, the shape coefficient of spheres (the idealized shape of glomeruli) and κ=1.1, the size distribution coefficient. Also, mesangial expansion was defined as a PAS-positive and nuclei-free area in the mesangium. Quantification of mesangial expansion was based on a minimum39of 10 glomeruli per section in a blinded fashion, under 400 × magnification (Zeiss AX10 microscope, Carl Zeiss Canada Ltd, Toronto, ON, Canada). The relative mesangial area was expressed as mesangial/glomerular surface area (%).
Cell culture
HK2 and HEK293T cells were obtained from ATCC (CRL-2,190 and CRL-3,216, respectively) and cultured according to their40specifications. Cells were stimulated with TM (Sigma-Aldrich) with or without 4-PBA (Sigma-Aldrich) as described in the legends. Conditionally immortalized human podocytes41 were obtained from Dr Moin Saleem, Bristol, UK. Cells were grown in RPMI-1,640 medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin antibiotics. Cells were cultured on rat tail collagen-I-coated plates and maintained at 33 °C (5% CO2, 90% humidity). Before experiments, cells were moved to a 37 °C incubator and cultured for at least 7 days to fully induce differentiation. Endotoxin-free human serum albumin was used (Sigma). Endotoxin levels in preparations were further determined by Associates of Cape Cod (Falmouth, MA, USA) and were found to be <2 endotoxin units per mg of protein.
Transfection
The Lonza’s Nucleofector Technology (Amaxa Basic Neucleofector kit for Primary Mammalian Epithelial Cells, Program T20) was used to transfect overexpression and shRNA knockdown constructs into HK2 cells with an efficiency of 80–90% based on GFP expression42. HEK293T cells were transfected using PolyJet transfection reagent (SignaGen Laboratories). Overexpression vector of RTN1A was obtained from Thermo Scientific and different clones of shRNA for RTN1 were obtained from Open Biosystems. The efficiency of overexpression and knockdown were confirmed by both western blot and real-time PCR. The expression constructs for WT-RTN1A-FLAG, MT-RTN1A-FLAG with deletion of C-terminal domain (amino acid 1–727), and RTN1C-FLAG were generated using a PCR-based mutagenesis method. To generate the human RTN1A-FLAG WT construct, we used the following primers:
5′- TGCTAGCCACCATGGCCGCGCCGGGGGATCCACAGCGAGCTCATCATCG -3′ and 5′- AGGTCGACATCGATTA TTTGTCATCGTCGTCCTTGTAGTCCAT GATATCCTCAGCGTGCCTCTTAGC -3′. The FLAG sequence is shown in italic, introduced restriction enzyme sites is in bold and stop code is in underline. The PCR products were purified and then ligated into pGEM-T easy Vector (Promega). After amplification, the DNA fragment was excised from the pGEM-T easy with NheI and NotI and then ligated into pEGFP-N1 (Clontech) at the same restriction enzyme sites. For MT-RTN1A, the primers used are: 5′- TTACTCGAGTTCCAACCAAAGTCCTG -3′ and 5′- TTTGATATCAGCCATGAGCAGCAGGGTCAG -3′. For cloning human RTN1C-FLAG, the primers used are 5′- TAGCTAGCCACCATGCAGGCCACTGCCGATTCCACCAAGATGGACT GTGTGTGGAGCAACTGGAAAAGTCAGGCTATTGACCTGTTGTATTGG -3′.
Lentiviral preparation and infection
HEK293T cells were transfected with either lentiviral plasmid expressing RTN1 shRNA sequence pGIPZ-shRTN1 (Open biosystems) or control scrambled sequence, pGIPZ-scramble, plus psPAX2 packaging plasmid and pMD2.G envelop plasmid using PolyJet transfection reagent according to the manufacturer’s protocol (SignaGen Laboratories). Forty-eight hours after transfection the lentiviral particles were harvested from HEK293T cell culture medium. Concentrated lentiviral particles were used to infect HK2 cells or podocytes.
Apoptosis analysis
Apoptosis was measured by flow cytometry using Annexin V-FITC apoptosis detection kit (BD Bioscience). Annexin V-FITC staining detects early stage apoptosis. Necrotic or late-stage apoptotic cells were labelled by propidium iodide. The number of cells labelled with Annexin V-FITC and propidium iodide was quantified using the FACS Caliber Flow cytometer and the data were analysed with CellQuest software (BD Biosciences). Caspase-3 activity was measured in HK2 cells using a Human Active Caspase-3 Immunoassay Kit (R&D Systems, Inc.) following the manufacturer’s protocol.
Real-time PCR
Samples were stored in RNAlater (Qiagen) solution at −80 °C until processing. Total RNA was extracted from isolated glomeruli or cultured podocytes using RNeasy Mini Kit (Qiagen). Superscript III First-Strand Synthesis SuperMix (Invitrogen) was used for reverse transcription of 1 μg of total RNA. PCR was performed using SYBR Green Master Mix (Applied Biosystems) and the Applied Biosystems 7500 Real-time PCR system. Intron-spanning primer sets selectively targeting mRNA and not genomic DNA sequence were designed using Primer-BLAST (NCBI) (Supplementary Table 6). The homogeneity and the size of PCR amplicons were confirmed by both melting curve analysis and agarose gel electrophoresis. Data were analysed by the 2−ΔΔCT method43 and presented as fold change relative to a control sample after normalization against the expression of a housekee** gene.
Western blot
Tissue or cells were lysed with a buffer containing 1% NP40, a protease and phosphatase inhibitor cocktail. After protein concentration determination, cell lysates were44subjected to western blot analysis using specific antibodies (Supplementary Table 7).
Immunoprecipitation
HK2 cells were transfected with FLAG-tagged WT-RTN1A, MT-RTN1A or RTN1C for 3 days. Cells were lysed with above lysis buffer and incubated with anti-PERK antibody (Cell Signaling) for overnight at 4 oC and the precipitated materials were used for western blot analysis using anti-FLAG antibody (Sigma). For co-IP with endogenous protein, HK2 cells were treated with tunicamycin at 25 ng ml−1 or dimethylsulphoxide as control for 24 h. Then, cells were lysed as above and incubated with anti-PERK antibody for IP and then western blot analysis with anti-RTN1A antibody as above.
Microarray studies
Affymetrix gene expression microarrays were performed at the Mount Sinai Institution Microarray Core Facility. The Affymetrix GeneChip Mouse Genome 430 2.0 Array was used to profile gene expression in the kidney cortex of Tg26 and WT mice37. One-way ANOVA was applied to the data set to identify the genes that were differentially expressed between the two groups. P values were corrected using Benjamini–Hochberg false discovery rate with a threshold of 0.05. Microarray data have been deposited in NCBI GEO under accession code GSE69074.
Immunostaining of kidney sections
Archival human kidney biopsies were collected at Jacobi Medical Center and Mount Sinai Hospital under protocols approved by their Institutional Review Boards. Biopsy samples included 5 cases of HIVAN, 5 MCD, 18 DN and 7 normal tissues from nephrectomy samples. Five samples were used to detect the average expression level of RTN1A in the specific kidney disease. More DN patients were selected from both Jacobi Medical Center and Mount Sinai Hospital for establishing a correlation between RTN1A expression and the severity of kidney disease in this patient population. We obtained only seven normal tissues from nephrectomy samples. Immunostaining was performed using specific primary antibodies (Supplementary Table 7) and biotinylated secondary antibodies (Vector Laboratories Inc.). Staining was revealed with avidin–peroxidase (VECTASTAIN Elite; Vector Laboratories Inc.). Slides were mounted in Aqua Poly/Mount (Polysciences Inc.) and photographed under an Olympus BX60 microscope with a digital camera. The following antibodies were used: RTN1A (Abcam). The extent of kidney staining in human biopsies was semi-quantitatively scored. The staining of each glomerulus was scored in a scale of 0–4 by two independent investigators (score 0: absence of specific staining; score 1: <25% area has specific staining for RTN1A; score 2: 25–50%; score 3: 50–75%; and score 4: >75%). The average score of an individual patient was calculated by adding of all scores from individual glomeruli divided by the number of glomeruli in the kidney section. The average score of RTN1A staining and eGFR or Scr from individual patients were used for calculation of correlation in patients with DN using the Pearson and Spearman method.
Statistical analysis
Data expressed as mean±s.d. (X±s.d.). The two-sided unpaired t-test was used to analyse data between the two groups after determination of data distributions and variance. The ANOVA with Bonferroni correction was used when more than two groups were present. Statistical significance was considered when P<0.05.
Additional information
Accession codes: Microarray data have been deposited in NCBI GEO under accession code GSE69074.
How to cite this article: Fan, Y. et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat. Commun. 6:7841 doi: 10.1038/ncomms8841 (2015).
Accession codes
Change history
22 October 2015
The original version of the Supplementary Information attached to this Article did not contain Supplementary Tables 1-7. The HTML has now been updated to include a corrected version of the Supplementary Information.
References
Coresh, J. et al. Prevalence of chronic kidney disease in the United States. JAMA 298, 2038–2047 (2007).
Lewis, E. J., Hunsicker, L. G., Bain, R. P. & Rohde, R. D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 329, 1456–1462 (1993).
Brenner, B. M. et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 345, 861–869 (2001).
de Zeeuw, D. Unmet need in renal protection--do we need a more comprehensive approach? Contrib. Nephrol. 171, 157–160 (2011).
Masliah, E. et al. Genetic deletion of Nogo/Rtn4 ameliorates behavioral and neuropathological outcomes in amyloid precursor protein transgenic mice. Neuroscience 169, 488–494 (2010).
Heath, J. E. et al. Widespread distribution of reticulon-3 in various neurodegenerative diseases. Neuropathology 30, 574–579 (2010).
Abdesselem, H., Shypitsyna, A., Solis, G. P., Bodrikov, V. & Stuermer, C. A. No Nogo66- and NgR-mediated inhibition of regenerating axons in the zebrafish optic nerve. J. Neurosci. 29, 15489–15498 (2009).
**ang, R. & Zhao, S. RTN3 inducing apoptosis is modulated by an adhesion protein CRELD1. Mol. Cell. Biochem. 331, 225–230 (2009).
Liu, Y. et al. Reticulon RTN2B regulates trafficking and function of neuronal glutamate transporter EAAC1. J. Biol. Chem. 283, 6561–6571 (2008).
Iwahashi, J. & Hamada, N. Human reticulon 1-A and 1-B interact with a medium chain of the AP-2 adaptor complex. Cell. Mol. Biol. (Noisy-le-grand) 49, Online Pub OL467–OL471 (2003).
Fazi, B. et al. Acetylation of RTN-1C regulates the induction of ER stress by the inhibition of HDAC activity in neuroectodermal tumors. Oncogene 28, 3814–3824 (2009).
Di Sano, F., Fazi, B., Tufi, R., Nardacci, R. & Piacentini, M. Reticulon-1C acts as a molecular switch between endoplasmic reticulum stress and genotoxic cell death pathway in human neuroblastoma cells. J. Neurochem. 102, 345–353 (2007).
Lu, T. C., He, J. C. & Klotman, P. Animal models of HIV-associated nephropathy. Curr. Opin. Nephrol. Hypertens. 15, 233–237 (2006).
Ikee, R. et al. Involvement of transglutaminase-2 in pathological changes in renal disease. Nephron. Clin. Pract. 105, c139–c146 (2007).
Bussolati, B., Collino, F. & Camussi, G. CD133+ cells as a therapeutic target for kidney diseases. Expert Opin. Ther. Targets 16, 157–165 (2012).
Woroniecka, K. I. et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 (2011).
Lindenmeyer, M. T. et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J. Am. Soc. Nephrol. 19, 2225–2236 (2008).
GrandPre, T., Nakamura, F., Vartanian, T. & Strittmatter, S. M. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444 (2000).
Chiurchiu, V., Maccarrone, M. & Orlacchio, A. The role of reticulons in neurodegenerative diseases. Neuromolecular Med. 16, 3–15 (2014).
Premsrirut, P. K. et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145, 145–158 (2011).
He, W. et al. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat. Med. 10, 959–965 (2004).
Montenegro, G. et al. Mutations in the ER-sha** protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 122, 538–544 (2012).
Cunard, R. & Sharma, K. The endoplasmic reticulum stress response and diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 300, F1054–F1061 (2011).
Morse, E. et al. TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. Am. J. Physiol. Renal. Physiol. 299, F965–F972 (2010).
Kimura, K., **, H., Ogawa, M. & Aoe, T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem. Biophys. Res. Commun. 366, 1048–1053 (2008).
Liu, G. et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem. Biophys. Res. Commun. 370, 651–656 (2008).
Wu, X., He, Y., **g, Y., Li, K. & Zhang, J. Albumin overload induces apoptosis in renal tubular epithelial cells through a CHOP-dependent pathway. OMICS 14, 61–73 (2010).
Cybulsky, A. V. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 77, 187–193 (2010).
Lee, E. K. et al. Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species. Nephron Exp. Nephrol. 121, e38–e48 (2012).
Wu, J. et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am. J. Pathol. 176, 2163–2176 (2010).
Inagi, R. et al. Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int. 68, 2639–2650 (2005).
Nakatani, Y. et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 280, 847–851 (2005).
Ozawa, K. et al. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes 54, 657–663 (2005).
Ozcan, U. et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 (2006).
Luo, Z. F. et al. Effects of 4-phenylbutyric acid on the process and development of diabetic nephropathy induced in rats by streptozotocin: regulation of endoplasmic reticulum stress-oxidative activation. Toxicol. Appl. Pharmacol. 246, 49–57 (2010).
Ratnam, K. K. et al. Role of the retinoic acid receptor-alpha in HIV-associated nephropathy. Kidney Int. 79, 624–634 (2011).
**, Y. et al. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat. Med. 18, 580–588 (2012).
Mallipattu, S. K. et al. Expression of HIV transgene aggravates kidney injury in diabetic mice. Kidney Int. 83, 626–634 (2013).
Zheng, F. et al. Combined AGE inhibition and ACEi decreases the progression of established diabetic nephropathy in B6 db/db mice. Kidney Int. 70, 507–514 (2006).
Rosenstiel, P. E. et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. 74, 1049–1058 (2008).
Saleem, M. A. et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 (2002).
Chuang, P. Y., Yu, Q., Fang, W., Uribarri, J. & He, J. C. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 72, 965–976 (2007).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
He, J. C. et al. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J. Clin. Invest. 114, 643–651 (2004).
Acknowledgements
J.C.H. is supported by VA Merit Award, NIH 1R01DK078897, NIH 1R01DK088541, NIH P01-DK-56492, Chinese 973 fund 2012CB517601; P.Y.C. is supported by NIH 1R01DK098126-01A1.
Author information
Contributions
J.C.H., K.L., P.Y.C., N.W. and W.J. designed the research project; Y.F., W.X., Z.L. and X.L. performed the experiments; B.J. and H.X. provided the archival human kidney samples; W.Z., C.W., Y.F. and W.X. analysed the data; and J.C.H., K.L., Y.F. and W.X. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Figures and Tables
Supplementary Figures 1-20 and Tables 1-7. (PDF 2107 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fan, Y., **ao, W., Li, Z. et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 6, 7841 (2015). https://doi.org/10.1038/ncomms8841
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8841
- Springer Nature Limited
This article is cited by
-
DDRGK1-mediated ER-phagy attenuates acute kidney injury through ER-stress and apoptosis
Cell Death & Disease (2024)
-
Erythropoietin mitigated thioacetamide-induced renal injury via JAK2/STAT5 and AMPK pathway
Scientific Reports (2023)
-
Soluble Klotho protects against glomerular injury through regulation of ER stress response
Communications Biology (2023)
-
PRE-084 ameliorated kidney injury by reducing endoplasmic reticulum stress in the rat model of adenine-induced chronic kidney disease
Molecular Biology Reports (2023)
-
Loss of RTN3 phenocopies chronic kidney disease and results in activation of the IGF2-JAK2 pathway in proximal tubular epithelial cells
Experimental & Molecular Medicine (2022)