

Abstract
Most small-molecule inhibitors of voltage-gated ion channels display poor subtype specificity because they bind to highly conserved residues located in the channel’s central cavity. Using a combined approach of scanning mutagenesis, electrophysiology, chemical ligand modification, chemical cross-linking, MS/MS-analyses and molecular modelling, we provide evidence for the binding site for adamantane derivatives and their putative access pathway in Kv7.1/KCNE1 channels. The adamantane compounds, exemplified by JNJ303, are highly potent gating modifiers that bind to fenestrations that become available when KCNE1 accessory subunits are bound to Kv7.1 channels. This mode of regulation by auxiliary subunits may facilitate the future development of potent and highly subtype-specific Kv channel inhibitors.
Similar content being viewed by others
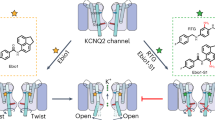

Introduction
Voltage-gated potassium (Kv) channels enable the rapid, selective and passive transport of potassium ions through cellular membranes that regulate physiological processes such as ion-coupled transport, hormone secretion, vesicle cycling and cell excitability. Dysfunction of Kv channels causes numerous inherited or acquired channelopathies, and these channels are under investigation as potential therapeutic targets for acquired disease such as cardiac arrhythmia, neurodegenerative diseases and diabetes1,2,3,4,5,6,7,8. Kv channel diversity is impressive and is enhanced by the large number of different α-subunits, alternative splicing, post-transcriptional modifications and coassembly of similar but not identical pore forming α-subunits and/or accessory β-subunits to form heteromeric channels9,10,11. β-subunits modify the pharmacology, subcellular localization, gating and ion selectivity of Kv channels12,13,14,15,16. For example, KCNE1 β-subunits coassemble with Kv7.1 α-subunits to increase current magnitude, slow the rate of activation and remove apparent inactivation gating17,18,19.
The design of small compound inhibitors of voltage-gated channels with high affinity and subtype specificity has been particularly challenging. Most known small-molecule pore blockers of Kv channels bind to specific residues that line the wall of the central cavity20,21,22,23,24. With few exceptions25,26, these crucial residues are conserved in most K+ channels, complicating the discovery and development of subtype-specific channel inhibitors. Highly potent and selective peptide inhibitors (for example, natural toxins) that bind to a site outside the central cavity (for example, to the outer vestibule) are of limited practical use as therapeutic agents because they require parenteral administration and often have serious undesirable side effects8,25,27. Investigating the molecular basis of drug binding is also hampered by complicating issues of allosteric effects and studies are often limited to investigating the effects of point mutations on functional measures of drug effects, without directly assessing the site of drug binding.
Here we use multiple complementary approaches to characterize the binding mode of adamantane derivatives that can explain why these compounds are potent inhibitors of Kv7.1/KCNE1 channels. In addition to a conventional mutagenesis-based investigation of drug effects, we have generated an adamantane analog with a cross-linking moiety that allows direct map** of its binding to specific channel peptide segments. Our findings suggest that these adamantanes bind with nanomolar affinity to fenestrations in the Kv7.1 channel that only form when the channel is in a complex with KCNE1 β-subunits. The mechanism of allosteric inhibition described here provides new opportunities for develo** small-molecule inhibitors of heteromeric channels with the desired properties of very-high affinity and specificity.
Results
KCNE1 induces sensitivity of Kv7.1 to inhibition by AC-1
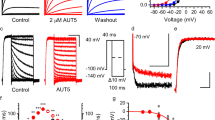
Compounds binding to the central cavity of Kv7.1 have been reported to act on both homomeric Kv7.1 and heteromeric Kv7.1/KCNE1 channels, albeit with varying potency20,21,Full size image
To directly test whether AC-1 preferentially accesses and stabilizes a non-conducting closed state, we applied the compound to oocytes clamped at −80 mV and analysed current inhibition by resuming pulsing after 4.5 min. Current was highly inhibited during the first post-rest pulse, suggesting that AC-1 can access its binding site when the channel is in a closed state (Fig. 2f). Conditioning subthreshold prepulses at 0.2 and 2 Hz also revealed use-dependent drug effects suggesting inhibition in preopen states (Fig. 2g). We also tested the onset of current inhibition during wash-in of AC-1 while holding oocytes at various membrane potentials. The onset of block was significantly faster with the more negative holding potentials, suggesting that the drug interacts preferentially with a closed/pre-open channel state (Supplementary Fig. 3).
Ala scanning of S4, S5, S6 and proximal C terminus of Kv7.1
Ala scanning was previously used to identify specific residues that form the putative binding sites for chromanol and benzodiazepine derivatives that are considered to be classical pore blockers of Kv7.1 channels20,21. Here we applied Ala scanning to multiple segments of the Kv7.1 α-subunit to identify potential AC-1 interaction sites in the extended pore region of Kv7.1/KCNE1 channels (Fig. 3a). In cases where mutation to Ala precluded channel function, alternative residues were introduced. This approach identified one residue in S4, two in S5, one residue at the base of the pore helix, and seven residues in S6, that markedly reduced AC-1 sensitivity when mutated (Fig. 3b, Supplementary Fig. 4, Table 1). The S6 residues identified by Ala scanning form a cluster in the channel structure, but do not all face the central cavity in three-dimensional (3D) models of open, closed34 or preopen states35. Instead, most of these crucial residues cluster around a ‘window’ formed uniquely in the preopen–closed state model of the Kv7.1/KCNE1 channel33 (Fig. 3c, magenta residues). This window structure resembles the fenestrations described for the NavAb channel and proposed to be a binding site for local anesthetics that inhibit Nav channels36,37. Docking of AC-1 to our previously published preopen–closed state model35 and subsequent MD-simulation was performed and indicated that AC-1 was stable in two positions along the longitudinal axis of the fenestrations (Fig. 3d). By contrast, in silico models of the closed and open states do not exhibit clear fenestrations (Supplementary Fig. 5) and thus, AC-1 cannot interact with this cavity in these channel states.

(a) Inhibition of wt and mutant Kv7.1/KCNE1 channels by 300 nM AC-1. Influence of amino acid exchange (yellow) on channel sensitivity to 300 nM AC-1 was investigated using alanine scanning combined with TEVC. Inhibition was determined as percent change in current amplitude at the end of a depolarizing test pulse (n=4–57, ±s.e.m.; one way analysis of variance, Dunnett’s post hoc test; ***P<0.001). (b) Cartoon of a single Kv7.1 channel subunit with scanned region in yellow. The circles indicate positions of mutations that significantly alter AC-1 sensitivity (key residues). Magenta filled circles indicate residues that face into the fenestration and blue filled circles mark residues that do not face fenestrations. (c) A preopen closed state model of Kv7.1/KCNE1 (Kv7.1 colored blue, KCNE1 colored yellow) with key residues highlighted in magenta (fenestration facing), blue non-fenestration facing and orange (fenestration facing in KCNE1). (d) Molecular docking of AC-1 to a homology model of Kv7.1/KCNE1 (ref. 33). AC-1 was positioned in close proximity to the amino acid residues identified by alanine scanning. AC-1 is located in a fenestration, which is only formed in the presence of KCNE1 (ref. 35).
Chemical modification of the AC-1 scaffold
We also investigated the functional effects of a panel of AC-1 analogs30 with chemical modifications at either end of the drug scaffold (Supplementary Table 1). Variation at position Y had little effect on inhibition. Introduction of substituents with variable volume and hydrophobicity at position X showed that small polar substituents reduce inhibition, whereas bulky substituents are tolerated and can increase apparent affinity (Fig. 4a). The potency of ACs was not dependent on hydrophobicity, quantified as logP using the Molinspiration logP calculator (Fig. 4b) or volume (Fig. 4c). Notably, results obtained with AC-9, an isobutyramide analog of the methylsulfonamide AC-2, indicate that the sulfonamide can be replaced with a carboxamide, and that relatively large lipophilic groups can be accommodated. These data clearly show that the AC-1 scaffold can be significantly expanded at either longitudinal end, suggesting that the AC binding site is not precisely length-delimited. We speculate that this SAR argues against the notion of a central cavity-constrained binding site, which would limit the dimensions by which drugs could be expanded while retaining effectiveness.

(a) Inhibitory effect of different AC derivatives (10 μM) on Kv7.1/KCNE1 channels. Chemical modifications at both ends of AC-1 were investigated as indicated. Inhibition was determined as percent change in current amplitude at the end of a depolarizing test pulse to +40 mV (n=3–5, ±s.e.m.). (b) Lack of correlation between inhibitory effect and hydrophobicity of AC compounds (n=3–5, ±s.e.m.). (c) Lack of correlation between inhibitory effect and volume of AC derivatives (n=3–5, ±s.e.m.). miLog P values and volume were calculated using Property Calculator (Molinspiration Cheminformatics).
Photoaffinity labelling approach to identify AC interactions
Interpretation of mutagenesis-based investigation of drug binding sites is often hampered by the possibility of secondary allosteric effects that impact drug binding or alter drug response with no change in binding affinity. Therefore, we complemented our mutagenesis and modelling findings by develo** a photoaffinity labelling (PAL)-based approach to directly identify regions of the Kv7.1/KCNE1 complex that interact with the AC compounds. We designed and synthesized an AC-9 analog with a photo-activatable cross-linking moiety that could covalently bind to the Kv7.1/KCNE1 channel complex (Fig. 5a, step 1–2). Labelled channel complexes were purified, and modified peptides were identified using MS/MS spectrometry (Fig. 5a, step 3–4). The diazirine substituted AC analog used for chemical cross linking was synthesized by coupling an NHS-diazirine to the amino group of AC-4 (Fig. 4) to generate AC-10 (Supplementary Fig. 6).

(a) Schematic view of the PAL-based approach to investigate the binding site of AC-1. (b) Concentration-response curve for AC-10, the UV-active diazirine derivate of AC-1. The inhibitory effect of AC-10 was determined in CHO cells stably expressing Kv7.1/KCNE1. Inhibition was determined as percent change in current amplitude at the end of the depolarizing test pulse to +40 mV (±s.e.m.). (c) A new cDNA-construct (Kv7.1myc-2A-KCNE1myc in pXOOM) allows for functional expression of myc-tagged Kv7.1 and KCNE1 in HEK293T cells. In this construct, cDNAs of Kv7.1 and KCNE1 are linked via the T2A peptide sequence, which mediates co-translational protein cleavage42. (d) Western blot analysis of affinity purified Kv7.1 and KCNE1 proteins. Kv7.1myc was purified using myc-agarose beads KCNE1 was purified using anti-KCNE1 coupled protein-A-sepharose beads (right). In both western blots, ‘+’ indicates transfected cells, ‘−’ indicates non-transfected control cells. Antibodies used for affinity purification are detected in ‘+’ and ‘−’. Additional protein bands proved the expression of Kv7.1 and KCNE1 proteins. It should be noted, that KCNE1, as a myc-tagged protein, should also be purified using the myc-agarose beads. Since the respective KCNE1 band is of comparable size to the anti-myc band, it is not possible to distinguish between them. Thus, KCNE1 expression was instead proven using the KCNE1 antibody. (e) Functional expression of Kv7.1myc-2A-KCNE1myc in HEK293T cells (scale bars indicate 1 nA, 1 s).
Diazirines can be activated by UV irradiation at 350 nm, and generate highly reactive carbene species (Supplementary Fig. 6) leading to covalent binding to all natural amino acids38,39. AC-10 inhibited Kv7.1/KCNE1 channels with an IC50 of 16 nM, indicating that the diazirine adduct does not weaken drug binding. AC-10 did not inhibit homomeric Kv7.1 channels (Fig. 5b, Supplementary Fig. 6c), Due to its very high apparent affinity, the effects of non UV-activated AC-10 could only be partially reversed by washout (40% inhibition after 38 min washout). After UV irradiation, current inhibition by AC-10 was mostly irreversible, suggesting that AC-10 was covalently bound and thus locked the channel in a non-conductive state (75% inhibition after 38 min washout, supplementary Fig. 6d). To achieve equal overexpression of subunits and to facilitate subsequent protein isolation, a bicistronic vector containing the cDNAs of c-myc tagged Kv7.1 and KCNE1 were linked via a T2A peptide sequence (Supplementary Fig. 7, Fig. 5c), a ∼20 amino acid sequence which mediates co-translational cleavage of polyproteins40,41,42. Transfection of HEK293T cells with this bicistronic vector leads to expression of Kv7.1myc and KCNE1myc proteins that were isolated via affinity chromatography and detected by Western blot analysis using a c-myc or anti-KCNE1 antibody (Fig. 5d). Functional expression of Kv7.1/KCNE1 channels in HEK293T cells was verified by patch clamp experiments (Fig. 5e). AC-10 was applied and covalently bound to channels overexpressed in HEK293T cells by UV irradiation. The tagged channel proteins were then purified on anti-myc columns, digested by trypsin, and the AC-10 modified peptides detected by MS/MS spectrometry (Fig. 6a,b, Supplementary Fig. 8 shows coverage of MS/MS). The only AC-10-modified peptide identified corresponds to the transmembrane helix of the β-subunit KCNE1 (42LEALYVLMVLGFFGFFTLGIMLSYIR67, Fig. 6c). Due to limited resolution, it was not possible to explicitly identify a single AC-10-bound amino acid residue. Another consideration is that some flexibility of the diazirine moiety may enable cross-linking at multiple positions in the identified peptide region. Nevertheless, no other labelled peptides (from Kv7.1 or KCNE1) were detected in three experimental trials, suggesting that there is no promiscuous cross-linking of the diazirine compound throughout the channel. Furthermore, when KCNE1 was expressed alone or in complex with Kv2.1, no KCNE1 modification was detected, indicating that both Kv7.1 and KCNE1 are required for AC-10 modification. Spectra for KCNE1 alone or in complex with Kv2.1 do not provide any evidence for modification by AC-10 (Supplementary Fig. 9). Rather, cross-linking with KCNE1 is very specific and reflects cross-linking taking place in a narrowly defined binding orientation that requires co-assembly with Kv7.1.

(a) Extracted ion chromatograms of the triply charged species of the KCNE1 peptide 42LEALYVLMVLGFFGFFTLGIMLSYIR67 with AC-10 modification (1163.28 m/z) identified across three replicate LC-MS/MS analyses. Isotope pattern of the [M+3H]3+ precursor of AC-10 modified peptide 42LEALYVLMVLGFFGFFTLGIMLSYIR67 (b) and corresponding MS/MS spectra with b- (red) and y-ion annotation (blue) (c). The characteristic marker ion of AC-10 (459.39m/z) is observed in the MS/MS spectrum. The consecutive y-ion series y2–y9 of respective amino acids is nicely displayed.
Cys scanning of the KCNE1 transmembrane segment
The β-subunit KCNE1 markedly increases sensitivity of Kv7.1 channels to classical pore blockers such as chromanol 293B and the benzodiazepine L-7 (refs 20, 21, 29). KCNE1 does not directly participate in binding of these inhibitors, but is thought to facilitate ligand binding to the α-subunit Kv7.1 via an allosteric mechanism29. In contrast, our PAL-based experiments indicate that the transmembrane segment of KCNE1 may be directly involved in binding of AC-1/AC-10. To further investigate whether specific KCNE1 residues interact with AC-1, cysteine substitutions of residues located in the central transmembrane region of KCNE1 (residues 52–58) were generated. Based on response of the mutant channels to 0.3 μM AC-1, Phe54 and Thr58 were identified as crucial residues for AC-1 binding (Fig. 7).

(a) Location of the transmembrane region of KCNE1 (KCNE1-TM) in a Kv7.1/KCNE1 channel complex. (b) Inhibition of Kv7.1/KCNE1 channel complexes by 300 nM AC-1. Single point mutations were introduced as indicated and inhibition was determined as percent change in current amplitude at the end of a depolarizing test pulse to +40 mV (n=3–5, ±s.e.m.; one-way analysis of variance, Dunnet’s post hoc test; ***P<0.001).
Constrained AC-1 docking
Results from PAL experiments, along with mutational scans of Kv7.1 and KCNE1, were used to facilitate a spatially restrained modelling of AC interaction with the Kv7.1/KCNE1 complex. To generate the model, AC-10 was introduced by swap**/adding atoms in the AC-1–Kv7.1/KCNE1 model shown in Fig. 3c. Next, the distance between the reactive AC-10 group and the side chain of Thr58 was constrained to a distance that allowed for covalent binding. The resulting model shows AC-10 bound in a position within the fenestrations of Kv7.1 (Fig. 8). We also performed 25 nsec simulations (all atoms mobile) on the AC-10–Kv7.1-KCNE1 model, and observed very stable binding in this conformation throughout the simulation. A parallel simulation on the identical Kv7.1-KCNE1 model without AC-10 showed significant fluctuation throughout the channel complex, indicating that AC-10 may stabilize the channel complex (Supplementary Fig. 10).

(a) AC-10 carbene docked by constraining the distance (2 ±1 Å) to the KCNE1 Thr58 side chain. The energy minimized model complex is shown. Kv7.1/KCNE1 was surface rendered, cut in the middle and colored blue, KCNE1 Thr58 is presented in ball-representation and CPK color coding with carbons in cyan. Compound AC-10 is shown in ball-representation and standard CPK color coding. K+-ions are shown as yellow spheres. (b) Close-up of AC-10 in its binding pocket. (c) Close-up representation of similar region, but with protein shown as ribbons (KCNE1 in magenta, Kv7.1 subunits in yellow green orange). (d) The AC binding site is formed by several residues that surround a tunnel-like structure (fenestration) that bridges the inner cavity (left) and the membrane (right). The surface lining the fenestration is hydrophobic, especially in the region around the adamantine of AC-1. (e) The specific ligand-receptor interactions determined using Discovery Studio 4.0. are: six alkyl interactions (pink), one alkyl-pi interaction (pink) and one carbon interaction (green).


