
Abstract
Background
Cancer cachexia is a severe wasting syndrome characterized by the progressive loss of lean body mass and systemic inflammation. Inhibiting the signaling of the transcription factor nuclear factor kappa B (NF-κB) largely prevents cancer-induced muscle wasting in murine models. We have previously shown the utility of Compound A, a highly selective novel NF-κB inhibitor that targets the IκB kinase complex, to provide clinical benefit in cancer-induced skeletal muscle and cardiac atrophy.
Methods
Using a metabolomics approach, we describe the changes found between cachectic and noncachectic gastrocnemius muscles before and after Compound A treatment at various doses.
Results
Of the 234 metabolites in the gastrocnemius, cachexia-induced changes in gastrocnemius metabolism reset the steady-state abundances of 42 metabolites (p < 0.05). These changes, not evenly distributed across biochemical categories, are concentrated in amino acids, peptides, carbohydrates and energetics intermediates, and lipids. The gastrocnemius glycolytic pathway is markedly altered—changes consistent with tumor Warburg physiology. This is the first account of a Warburg effect that is not exclusively restricted to cancer cells or rapidly proliferating nonmalignant cells. Cachectic gastrocnemius also displays tricarboxylic acid cycle disruptions, signs of oxidative stress, and impaired redox homeostasis. Compound A only partially rescues the phenotype of the cachectic gastrocnemius, failing to restore the gastrocnemius’ baseline metabolic profile.
Conclusions
The findings in the present manuscript enumerate the metabolic consequences of cachexia in the gastrocnemius and demonstrate that NF-kB targeted treatment only partly rescues the cachectic metabolic phenotype. These data strengthen the previous findings from metabolomic characterization of serum in cachectic animals, suggesting that many of the metabolic alterations observed in the blood originate in the diseased muscle. These findings provide significant insight into the complex pathophysiology of cancer cachexia and provide objective criteria for evaluating future therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cancer cachexia is a complex metabolic and nutritional syndrome characterized by weight loss, loss of muscle and adipose tissue, weakness affecting functional status, impaired immune system, and alterations in carbohydrate, lipid, and protein metabolism [1–3]. Unlike starvation, where lean mass is preserved, cancer cachexia is a progressive wasting disorder with loss of skeletal and visceral muscle, with or without fat, frequently associated with inflammation and insulin resistance [4, 5]. The syndrome is correlated with increasing morbidity and mortality, along with significant impairment of quality of life and response to treatment [1, 6]. In fact, cancer cachexia is estimated to occur in 50–80 % of cancer patients and accounts for 20–30 % of all cancer-related deaths [7].
The C26 murine undifferentiated colon carcinoma model of cancer cachexia has been used to elucidate the mechanisms of the syndrome, which has been found to closely parallel human disease [8–10]. Recent studies using this model have identified a role for tumor release of proinflammatory cytokines such as IL-1, IL-6, and TNF-α in the mechanism of striated muscle loss [11]. These cytokines further activate transcription factor nuclear factor kappa B (NF-κB), which upregulates the ubiquitin-dependent degradation of the sarcomere leading to muscle atrophy [12]. Our and others’ recent work has shown that pharmacological and genetic inhibition of NF-κB is protective against cancer cachexia-induced muscle atrophy [13–15]. Compound A in particular is a highly selective and competitive NF-κB inhibitor that targets the upstream NF-κB regulator IκB kinase complex (IKK-β); this ensures that only acute increases in NF-κB, and not basal NF-κB activities, are affected [16]. There is evidence in the literature that compound A induces resistance to skeletal muscle injury [17] and is also cardioprotective against cancer cachexia-induced cardiac atrophy and systolic dysfunction [14].
While the mechanisms underlying muscle wasting in cancer cachexia are being investigated, little is known about the impact of this wasting on muscle properties including muscle function, but also on its metabolic properties [18]. Recently, metabolomics, or the systematic study of the small-molecule metabolite profiles that specific cellular processes leave behind, has been used to yield a better understanding of cancer cachexia. For instance, we have shown that cancer cachectic mice sera have a unique metabolic fingerprint that confirms cancer cachexia as a distinct entity from a healthy state, other wasting disorders, and cancer itself [19]. Similar studies are aiming at discovering specific biomarkers of cancer cachexia. In the present study, we sought to identify changes in cachectic murine gastrocnemius muscle samples and the effect of Compound A treatment, using multiple-platform mass spectrometry and nontargeted biochemical profiling.
In this study, we report multiple changes in the cachectic gastrocnemius that are partially rescued with Compound A treatment. These changes give insight into the mechanisms of cancer cachexia. One change in particular, the Warburg-like metabolic signature in the cachectic skeletal muscle, is to our knowledge, the first evidence of the existence of such activity in cells other than cancer cells or rapidly proliferating noncancerous cells.
2 Material and methods
2.1 Cell line
The cachexia-inducing transplantable C26 undifferentiated colon carcinoma cell line was a kind gift from Dr. Denis Guttridge (Columbus, OH, USA). The cells were maintained as monolayers in culture flasks at 37 °C with 5 % CO2 in culture medium which consisted of Roswell Park Memorial Institute 1640 (Gibco, Rockville, MD, USA) supplemented with 5 % fetal bovine serum (Hyclone, Logan, UT, USA) and 1 % penicillin/streptomycin (Gibco). Immediately before injection, cells were trypsinized at approximately 80 % confluence, washed, and suspended in phosphate-buffered saline at a concentration of 5 × 106 cells/ml.
2.2 Experimental protocol
Thirty-three male BALB/c mice (Charles River Laboratories, Wilmington, MA, USA) were housed under conventional conditions with constant temperature and humidity and were maintained on a 12:12-h dark–light cycle. Mice were housed in five animals per cage and were allowed free access to standard diet and water. Treatment of mice was in accordance with the guidelines of the Institutional Animal Care and Use Committee. After being allowed to acclimate to their new environment for 3 days before beginning the study, the mice aged 43–63 days (weight, 22–24 g) were randomly divided into five groups: healthy control mice, tumor-bearing mice (dimethyl sulfoxide [DMSO] sham), and tumor-bearing plus the anti-inflammatory drug Compound A (2, 5, or 10 mg/kg). Body weight, tumor volume, and food consumption were measured every other day from inoculation to completion of the study. Tumor growth was assessed using calipers in two orthogonal dimensions (x = shortest diameter, y = longest diameter). Tumor volume was calculated using the following formula: \( V = \left( {{\pi \left/ {6} \right.}} \right)*{x^2}y \).
On day 0, the mice in tumor-bearing groups were injected subcutaneously in the right flank with 100 μl (approximately 500,000 cells) of C26 undifferentiated colon carcinoma cells. Beginning on day 6, when tumors were first noticed, untreated tumor-bearing mice received daily single intraperitoneal injections of sterile DMSO, while treated tumor-bearing mice received injections of Compound A (a highly selective novel NF-κB inhibitor with an anti-inflammatory role) at 2, 5, or 10 mg/kg. On day 17, when the majority of tumor-bearing mice had a significant tumor burden with clinical signs of cachexia, all mice were euthanized. One animal in the tumor-bearing group was sacrificed early due to tumor volume in excess of IUCAC guidelines. A final total body weight measurement was obtained. Final body weight was calculated by subtracting excised tumor weight from the last total body weight measurement (weight of mice with tumors intact). Tumor weight was measured from the gross specimens harvested at the completion of the study. Hind limbs were weighed separately as a measurement of lean body mass. Tumors were resected, measured, and weighed, and a total carcass weight (total body weight minus the tumor weight) was calculated. Gastrocnemius muscles and tumors were excised from each animal immediately after sacrifice, snap frozen in liquid nitrogen, and stored at −80 °C.
2.3 Metabolomic analysis
Biochemical profiling was performed using multiple platform (ultra high-pressure liquid chromatography (UHPLC) and gas chromatography (GC)) mass spectrometry technology, as described [20–23]. Briefly, a broad array of small molecule metabolites, irrespective of class (e.g., amino acids, lipids, carbohydrates), was examined to measure biochemical changes within samples. The nontargeted process used single sample extraction followed by protein precipitation to recover a diverse range of molecules (e.g., polar and hydrophobic).
2.4 Metabolite identification
Metabolites were identified by automated comparison and spectra fitting to a chemical standard library of experimentally derived spectra as previously described [20–22]. Identification of known chemical entities was based on comparison with library entries of purified authentic chemical standards. Of the metabolites, 243 were identified in gastrocnemius muscles using this global biochemical profiling analysis.
2.5 Sample preparation
Frozen gastrocnemius tissues (100 mg) were thawed on ice, combined with 0.5 mL of water and disaggregated using a bead mill homogenizer (1,350 strokes/min for 5 min, Geno/Grinder 200 (Glen Mills Inc., Clifton, NJ, USA)). An aliquot of each sample was extracted with methanol (0.4 mL) containing the recovery standards as previously reported [20]. After extraction, the sample was centrifuged and supernatant removed using the MicroLab STAR® robotics system. The extract supernatant was split into four equal aliquots (0.11 mL each): two for UHPLC/mass spectrometry (MS), one for GC/MS, and one reserve aliquot. Aliquots were placed on a TurboVap® (Zymark) to remove solvent, and dried under vacuum overnight. Samples were maintained at 4 °C throughout the extraction process. For UHPLC/MS analysis, extract aliquots were reconstituted in either 0.1 % formic acid for positive ion UHPLC/MS or 6.5 mM ammonium bicarbonate pH 8.0 for negative ion UHPLC/MS. For GC/MS analysis, aliquots were derivatized using equal parts of N,O-bistrimethylsilyl-trifluoroacetamide and a solvent mixture of acetonitrile/dichloromethane/cyclohexane (541) with 5 % triethylamine at 60 °C for 1 h. The derivatization mixture also contained a series of alkyl benzenes for use as retention time markers.
2.6 GC/MS and UHPLC/MS/MS analysis
UHPLC/MS/MS analysis was performed on a Waters Acquity UHPLC (Waters Corporation, Milford, MA, USA) coupled to an linear trap quadrupole (LTQ) mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) equipped with an electrospray ionization source. Two separate UHPLC injections were performed on each sample: one optimized for positive ions and one for negative ions. The mobile phase for positive ion analysis consisted of 0.1 % formic acid in H2O (solvent A) and 0.1 % formic acid in methanol (solvent B), while the mobile phase for negative ion analysis consisted of 6.5 mM ammonium bicarbonate, pH 8.0 (solvent A) and 6.5 mM ammonium bicarbonate in 95 % methanol (solvent B). Acidic extracts were evaluated for positive ions while basic extracts were evaluated for negative ions in independent injections using dedicated chromatographic columns (2.1 × 100 mm BEH C18 with 1.7 μm particle, Waters) maintained at 40 °C. The LTQ alternated between full-scan mass spectra (99–1,000 m/z) and data dependent MS/MS scans, which used dynamic exclusion.
The derivatized samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single–quadrupole MS operated at unit mass resolving power. Separation occurred through a temperature ramp (60–340 °C) using helium as a carrier gas on a GC column (20 m × 0.18 mm with 0.18 μm film phase consisting of 5 % phenyldimethyl silicone). The MS employed electron impact ionization with a 50–750 amu scan range and was tuned and calibrated daily for mass resolution and mass accuracy.
2.7 Data normalization
All samples were analyzed on a single day. For each metabolite, the raw area counts were divided by its median value. Missing values were assumed to result from areas falling below limits of detection. For each metabolite, missing values were imputed with its observed minimum after the normalization step.
2.8 Data extraction and quality assurance
Peaks were identified using Metabolon’s proprietary peak integration software and component parts were stored in a separate and specifically designed complex data structure [24].
Process quality was evaluated by pooling an aliquot (10 μL) of additional samples, each experimental sample together to serve as a technical replicate for duration of the run. This technical replicate sample was injected throughout the platform run day, allowing variability in quantitation of detected biochemicals to be monitored. With this monitoring, a metric on overall process variability was assigned for the platform’s performance (see “Results” section).
The median relative standard deviation (MRSD), a quality assurance metric of quantification and measure of instrument variability, was determined to be 8 % for a panel of 30 internal standards [25]. Overall process variability (i.e., extraction, recovery, resuspension, and instrument performance) for endogenous biochemicals within technical replicate samples was calculated to be 15 % MRSD. These standard deviation values reflected acceptable levels of variability for overall process and instrumentation of the analytical platform.
2.9 Statistical analysis
Statistical analyses were performed using JMP (JMP, Version 8. SAS Institute Inc., Cary, NC, USA, 1989–2009) and “R” (http://cran.r-project.org/).
3 Results
3.1 Metabolic derangements in the cachectic gastrocnemius
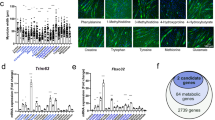
Of the 234 metabolites from all major biochemical categories that were found in the gastrocnemius, cachexia-induced changes in gastrocnemius metabolism resulted in altering the steady-state abundances of 42 of these metabolites, either increasing or decreasing them compared to controls (p < 0.05, FDR = 0.26; Fig. 1 and Electronic supplementary material (ESM) 1). These changes were not evenly distributed across biochemical categories, but instead were concentrated in amino acids and peptides (15), carbohydrates and energetics intermediates (14), and lipids (especially medium chain fatty acids and polar lipid head groups, 12).

Heat map of metabolite abundance profiles showing metabolic derangements in cachectic gastrocnemius. Of the 234 metabolites in the gastrocnemius, cachexia-induced changes reset the steady-state abundances of 42 metabolites (p < 0.05). These changes are not evenly distributed across biochemical categories. The gastrocnemius glycolytic pathway is markedly altered—changes consistent with tumor Warburg physiology. This is the first account of a Warburg effect that is not exclusively restricted to cancer cells or rapidly proliferating nonmalignant cells
Further consequences of cachexia in the gastrocnemius are biochemical signatures of oxidative and nitrogen stresses. The oxidative stress can be observed in the depletion of antioxidant peptides glutathione, anserine, and carnosine from diseased muscle (Fig. 2a). A further finding supporting the increased oxidative stress or impaired redox homeostasis in the cachectic muscle was the observation that the mannitol/mannose ratio shifted toward the oxidized form (mannitol/mannose ratio in the controls is 2.4 ± 1.1 compared to 0.56 ± 0.25 in the cachectic condition (p < 0.05).). Cachectic gastrocnemius also displays evidence of increased nitrogen stress and glutamine reduction with increased levels of the urea cycle intermediates, ornithine and arginine, in cachectic gastrocnemius samples (Fig. 2b).

Boxplots illustrating the major observed alterations in oxidative and nitrogen stress in the cachectic gastrocnemius. a Markers associated with redox state, b cationic amino acids
The gastrocnemius glycolytic pathway was markedly altered during cachexia. Specifically, there was a dramatic increase in the abundance of hexose-6-phosphates, including glucose-6-phosphate, fructose-6-phosphate, and mannose-6-phosphate (Fig. 3a). At the same time, there was a marked decrease in the abundance of the phosphorylated, 3-carbon glycolytic fragments 3-phosphoglycerate, 2-phosphoglycerate, and phosphoenolpyruvate (Fig. 3b). Consistent with the rebalancing of glycolytic intermediates in the cachectic gastrocnemius were findings that metabolites related to glycolysis were also altered in cachexia. Consistent with the decreased abundance of 3-carbon glycolytic fragments were reduced abundances of the amino acids that are made from 3-carbon glycolytic fragments (serine and glycine from 3-phosphoglycerate, and alanine from pyruvate; Fig. 3c). The lower amount of 3-carbon glycolytic intermediates was also reflected in a lower abundance of citrate and malate, key intermediates in the tricarboxylic acid (TCA) cycle (Fig. 3d). Although those two TCA cycle intermediates were decreased, there was a significant increase in the abundance of succinylcarnitine, a surrogate for the measurement of succinyl-CoA (Fig. 3d).

Box plots illustrating the major observed alterations to the glycolytic and TCA pathways in the cachectic gastrocnemius. a Elevation of hexose phosphates, b reduction in 3-carbon glycolytic intermediates, c reduction of amino acids that are made from 3-carbon glycolytic fragments, and d alteration in the abundance patterns of TCA cycle intermediates
3.2 Anti-inflammatory treatment partially rescues the metabolic phenotype of the cachectic gastrocnemius
Treatment with the anti-inflammatory agent Compound A had a modest effect on the biochemistry of the cachectic gastrocnemius. Of the 42 disease-associated changes, 11 were altered by treatment (ESM 1). Compound A treatment restored the abundance of the antioxidant histidine dipeptides carnosine and anserine to control levels (Fig. 4a). Also, galactitol, the product of aldose reductase-mediated reduction of galactose was partially rescued (Fig. 4a). Two of the polar lipid head groups, ethanolamine and inositol-1-phosphate also recovered to control levels with treatment (Fig. 4b). Compound A treatment resulted in partial recovery of energetics pathway metabolite levels; the glycolytic hexose phosphates (glucose-6-phosphate and fructose-6-phosphate) and citrate were brought back to their control levels (Fig. 4c).

Box plots illustrating the major observed alterations in the cachectic gastrocnemius with or without Compound A treatment compared to control. Compound A treatment effect on a markers associated with redox state, b polar lipid head groups, and c energetics pathway compounds
4 Discussion
In this study, mice were inoculated with the C-26 murine undifferentiated colon carcinoma, an established model that results in muscle wasting that mimics clinical cancer-related cachexia. This tumor cell line was previously shown to cause significant loss of mass in gastrocnemius and extensor digitorum longus muscles in mice without significant effects on food intake [11, 14, 15, 19]. Thus, any muscle wasting associated with this tumor cannot be attributed to anorexia and altered energy intake.
Metabolomic analysis of gastrocnemius muscles from animals with established cachexia revealed two predominant effects of cancer cachexia: increased oxidative stress and a disease-associated shift in glucose utilization through a high rate of glycolysis. The latter is similar to the physiology seen in tumor-related “Warburg effect.” An overview of the alterations in both central carbon and nitrogen metabolism in the cachectic gastrocnemius is shown in Fig. 5. The metabolic evidence of the redox imbalance in the cachectic gastrocnemius was the depletion of glutathione, the reduced abundance of antioxidant histidine dipeptides, and a shift in the ratios of redox-related metabolites toward their oxidized forms.

Overview of alterations in both central carbon and nitrogen metabolism in the cachectic gastrocnemius. Pronounced alterations occurred in both central carbon metabolism and nitrogen metabolism in the cachectic gastrocnemius. Metabolites immediately downstream of glucose, including fructose-6-phosphate, mannose-6-phosphate and mannose accumulated while the 3 carbon glycolytic fragments and metabolites derived from those compounds (including the 3-carbon amino acids serine and alanine and TCA cycle intermediates) decreased. At the same time, the urea cycle intermediates arginine and ornithine significantly increased
Oxidative stress derives from an imbalance between production and neutralization of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) [26, 27]. Indicators of ROS and RNS insults in skeletal muscle have been previously reported to be increased both in animal models of cancer cachexia [28, 29] and in cancer patients [30]. The ratio of oxidized glutathione to reduced glutathione, as measured in cachectic gastrocnemius samples, has been shown to be significantly increased, along with a decrease of the antioxidant enzyme superoxide dismutase activity [26].
ROS is known to cause damage to cellular components, organelles, membranes, and individual proteins; many cell types respond to this type of damage by inducing autophagy, a cellular recycling program whereby damaged macromolecular complexes are broken down into their component parts for reuse. While there is considerable evidence supporting autophagy in cardiac muscle, the data are not clearcut in skeletal muscle. Recently published electron micrographs of skeletal muscle from cachectic mice document electron lucent regions and swelling—ultrastructural findings that are suggestive of autophagy [31]. One of the autophagy triggers is a nutrient sensor signaling pathway and elevated glutamine abundance has been found to stimulate that pathway. However, in this study, glutamine was found to decrease in the cachectic gastrocnemius.
Several investigators have recently found that cancer cells can cause metabolic alterations in their surrounding stromal cells, effectively catabolizing the resources within the stroma to provide high-quality fuel sources for the tumor [32]. In many systems, cancer-associated fibroblasts have now been shown to have increased autophagy and actively secrete glutamine for the adjacent tumor [33]. These data suggest that cachexia-inducing tumors may be reprogramming the metabolism of skeletal muscle to catabolize their components for high-quality fuel sources, even though the tumors are distant from the muscle. Further work will be necessary to synthesize the present manuscript with these findings and determine the role that autophagy may play in this process.
ROS also leads to upregulation of the ubiquitin/proteasome pathway through NF-κβ activation accelerating protein degradation and muscle mass loss [28, 34, 35]. Alterations in muscle energetics are also intimately linked with tissue redox status and the pattern of glucose utilization intermediates in the glycolytic pathway was significantly altered in the cachectic gastrocnemius. The observed pattern, an increase in hexose phosphates with a concomitant decrease in 3-carbon glycolytic fragments, is similar to that seen with the “Warburg effect” that has been extensively studied in tumor metabolism. This study provides the first evidence at the possibility of a Warburg-like metabolism in skeletal muscle. Further studies are necessary in order to determine whether these findings truly represent a Warburg-like metabolism or are due to altered cellularity in the muscle at this point in the disease process.
The present manuscript illustrates steady-state metabolism in the cachectic gastrocnemius at a single time point in disease progression. At this time point, there was already significant muscle mass lost and damage to the molecular components of the tissue has accumulated. Further work, such as a time course examining both skeletal muscle ultrastructure and metabolism, could help to clarify the roles of autophagy and metabolic reprogramming in skeletal muscle that appear to occur in the presence of a cachexia-inducing tumor. As opposed to normal differentiated cells, which rely primarily on mitochondrial oxidative phosphorylation to generate the energy needed for cellular processes, most cancer cells instead rely on aerobic glycolysis [36]. This phenomenon enables rapidly dividing tumor cells to generate essential biosynthetic building blocks such as nucleic acids, amino acids, and lipids from glycolytic intermediates to permit growth and duplication of cellular components during division [37]. One consequence of the rebalancing of glycolytic intermediates is the apparent shunting of metabolism away from the TCA cycle and consequently oxidative phosphorylation.
It is also noteworthy that a potential rationale for the switch to a Warburg-like effect might be driven by skeletal muscle satellite cells. In fact, there is evidence of increased satellite cell proliferation during times of fiber wasting [38]. These cells might acquire a Warburg-like metabolism to support rapid proliferation [39].
A further defect in cachectic muscle samples affects the TCA cycle. Intermediates, such as citrate and malate were also markedly reduced, whereas succinylcarnitine was increased. These changes in TCA cycle intermediates suggest that during cachexia there is less carbon flowing through the TCA cycle and further suggest that the defect lies between succinyl-CoA and malate. Three enzymes of the TCA cycle are known to be sensitive to oxidative stress; namely aconitase, α-ketoglutarate dehydrogenase, and succinate dehydrogenase, with varying sensitivities and consequences of their impaired function [40, 41]. The reduced abundance of 3-carbon glycolytic intermediates and citrate suggest that less substrate is entering the TCA cycle. The increased abundance of succinylcarnitine in these same tissues indicates that there is a defect in complex II activity that contains succinate dehydrogenase and couples the TCA cycle and oxidative phosphorylation.
Chronic inflammation in the context of cancer, such as with cachexia, is mediated by proinflammatory cytokines that are regulated by NF-κB, making the latter an appealing target for therapeutic intervention [42]. NF-κB signaling is controlled by the inhibitor of IKK, a critical catalytic subunit of which is IKK-β. Compound A is a potent and selective IKK-β inhibitor and acts as a promising anti-inflammatory in vitro and in vivo [16]. Unlike proteasome inhibitors which block basal NF-κB activity [43] resulting in significant side effects and even lethality in humans [44], Compound A is focused on stress-induced NF-κB activation, allowing sufficient basal NF-κB activity to prevent apoptosis while still being sufficient to limit the inflammatory activity of NF-κB [16]. Therefore, we hypothesized that Compound A treatment would protect the gastrocnemius from cancer-induced cachexia. What we observed was that the cachexia-induced changes in the gastrocnemius were only partially corrected with Compound A treatment.
Compound A is given systemically, allowing NF-κB to be inhibited at the level of the heart, skeletal muscle, or other organs. The effectiveness of NF-κB inhibition due to Compound A treatment has been previously demonstrated in the cardiac muscles of the same mice used in this experiment [14]. NF-κB inhibition was determined using enzyme-linked immunosorbent assay for NF-κB activity, but also with the expression of Nfkbia, a gene regulated by NF-κB, as another surrogate of NF-κB activity.
Among the changes affected by Compound A treatment is the tissue redox environment of the gastrocnemius as evidenced by the renormalization of anti-inflammatory histidine dipeptides and polar lipid head groups. Also, galactitol, the product of aldose reductase-mediated reduction of galactose was partially rescued, suggesting that there was some NADPH available to mediate that reaction (as opposed to being utilized to facilitate glutathione recycling). It is well known that at least some of the effects exerted by oxidative stress are mediated by activation of the transcription factor NF-κB [45]. NF-κB activation in cancer cachexia has been shown in both murine models [15] and human cancer patients [46]. Inhibition of NF-κB by Compound A can explain the partial restoration of the normal oxidative environment.
5 Conclusion
Cancer cachexia has multiple effects on the gastrocnemius including increased oxidative stress and impaired redox homeostasis, changed pattern of glycolytic metabolite abundance consistent with tumor Warburg physiology, decreased carbon flow through the TCA cycle, and reduced abundance of amino acids. The failure to restore the baseline metabolic profile of the gastrocnemius with this treatment, especially the 3-carbon intermediates of glycolysis and amino acids, suggests that NF-κB inhibition does not address the root of the cachectic phenotype, but rather intervenes with significant symptomology.
References
Morley JE, Thomas DR, Wilson MM. Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr. 2006;83:735–43.
Tisdale MJ. Cancer cachexia. Langenbecks Arch Surg. 2004;389:299–305.
MacDonald N, Easson AM, Mazurak VC, Dunn GP, Baracos VE. Understanding and managing cancer cachexia. J Am Coll Surg. 2003;197:143–61.
Bennani-Baiti N, Walsh D. What is cancer anorexia-cachexia syndrome? A historical perspective. J R Coll Physicians Edinb. 2009;39:257–62.
Tisdale MJ. Cancer cachexia. Curr Opin Gastroenterol. 2010;26:146–51.
Tan BH, Fearon KC. Cachexia: prevalence and impact in medicine. Curr Opin Clin Nutr Metab Care. 2008;11:400–7.
Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–71.
Aulino P, Berardi E, Cardillo VM, Rizzuto E, Perniconi B, Ramina C, et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer. 2010;10:363.
Di Marco S, Cammas A, Lian XJ, Kovacs EN, Ma JF, Hall DT, et al. The translation inhibitor pateamine A prevents cachexia-induced muscle wasting in mice. Nat Commun. 2012;3:896.
Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One. 2011;6:e22538.
Fujita J, Tsu**aka T, Yano M, Ebisui C, Saito H, Katsume A, et al. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. International journal of cancer Journal international du cancer. 1996;68:637–43.
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
Van Gammeren D, Damrauer JS, Jackman RW, Kandarian SC. The IkappaB kinases IKKalpha and IKKbeta are necessary and sufficient for skeletal muscle atrophy. FASEB J. 2009;23:362–70.
Wysong A, Couch M, Shadfar S, Li L, Rodriguez JE, Asher S, et al. NF-kappaB inhibition protects against tumor-induced cardiac atrophy in vivo. Am J Pathol. 2011;178:1059–68.
Shadfar S, Couch ME, McKinney KA, Weinstein LJ, Yin X, Rodriguez JE, et al. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr Cancer. 2011;63:749–62.
Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol. 2005;145:178–92.
Tanaka K, Kiyosawa N, Honda K, Sharyo S, Ito K, Teranishi M, et al. Resistance to the skeletal muscle injury expressed by repeated treatment with compound A that has HMG-CoA reductase inhibitory activity. J Toxicol Sci. 2007;32:9–18.
Diffee GM, Kalfas K, Al-Majid S, McCarthy DO. Altered expression of skeletal muscle myosin isoforms in cancer cachexia. Am J Physiol Cell Physiol. 2002;283:C1376–82.
O’Connell T, Ardeshirpour F, Asher S, Winnike J, Yin X, George J, et al. Metabolomic analysis of cancer cachexia reveals distinct lipid and glucose alterations. Metabolomics. 2008;4:216–25.
Boudonck KJ, Mitchell MW, Nemet L, Keresztes L, Nyska A, Shinar D, et al. Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol Pathol. 2009;37:280–92.
Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, et al. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9:383–97.
Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–67.
Sha W, da Costa KA, Fischer LM, Milburn MV, Lawton KA, Berger A, et al. Metabolomic profiling can predict which humans will develop liver dysfunction when deprived of dietary choline. FASEB J. 2010;24:2962–75.
Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminformatics. 2010;2:9.
Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, et al. Alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS One. 2010;5:e10883.
Mastrocola R, Reffo P, Penna F, Tomasinelli CE, Boccuzzi G, Baccino FM, et al. Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radic Biol Med. 2008;44:584–93.
Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35:411–29.
Gomes-Marcondes MC, Tisdale MJ. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002;180:69–74.
Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005;579:1646–52.
Murr C, Fuith LC, Widner B, Wirleitner B, Baier-Bitterlich G, Fuchs D. Increased neopterin concentrations in patients with cancer: indicator of oxidative stress? Anticancer Res. 1999;19:1721–8.
Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, et al. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging. 2012;4:133–43.
Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16.
Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther. 2011;12:1085–97.
Reid MB, Li YP. Cytokines and oxidative signalling in skeletal muscle. Acta Physiol Scand. 2001;171:225–32.
van Wessel T, de Haan A, van der Laarse WJ, Jaspers RT. The muscle fiber type-fiber size paradox: hypertrophy or oxidative metabolism? Eur J Appl Physiol. 2010;110:665–94.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY). 2009;324:1029–33.
Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–7.
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44.
Lopez-Lazaro M. The Warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anti Cancer Agents Med Chem. 2008;8:305–12.
Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–45.
Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–9.
Gupta SC, Kim JH, Kannappan R, Reuter S, Dougherty PM, Aggarwal BB. Role of nuclear factor kappaB-mediated inflammatory pathways in cancer-related symptoms and their regulation by nutritional agents. Exp Biol Med (Maywood). 2011;236:658–71.
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–47.
Willis MS, Schisler JC, Patterson C. Appetite for destruction: E3 ubiquitin-ligase protection in cardiac disease. Future Cardiol. 2008;4:65–75.
Aragno M, Mastrocola R, Brignardello E, Catalano M, Robino G, Manti R, et al. Dehydroepiandrosterone modulates nuclear factor-kappaB activation in hippocampus of diabetic rats. Endocrinology. 2002;143:3250–8.
Rhoads MG, Kandarian SC, Pacelli F, Doglietto GB, Bossola M. Expression of NF-kappaB and IkappaB proteins in skeletal muscle of gastric cancer patients. Eur J Cancer. 2010;46:191–7.
Acknowledgments
Grant supports are from J. Walter Juckett postdoctoral fellowship (HD), Howard Hughes Medical Institute Research training fellowship (AW), University of North Carolina Program in Translational Science (MC), the American Heart Association Scientist Development (MW), and the National Heart, Lung, and Blood Institute grant R01HL104129 (MW). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle 2010;1:7–8 (von Haehling S, Morley JE, Coats AJ and Anker SD).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
XLSX 26 kb
About this article
Cite this article
Der-Torossian, H., Wysong, A., Shadfar, S. et al. Metabolic derangements in the gastrocnemius and the effect of Compound A therapy in a murine model of cancer cachexia. J Cachexia Sarcopenia Muscle 4, 145–155 (2013). https://doi.org/10.1007/s13539-012-0101-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-012-0101-7