
Abstract
Aquaporin-4 (AQP4)-IgG seropositive neuromyelitis optica spectrum disorders (AQP4-IgG seropositive NMOSD) and myelin oligodendrocyte glycoprotein (MOG)–IgG-associated disease (MOGAD) are inflammatory demyelinating disorders distinct from each other and from multiple sclerosis (MS).While anti-CD20 treatments can be used to treat MS and AQP4-IgG seropositive NMOSD, some MS medications are ineffective or could exacerbate AQP4-IgG seropositive NMOSD including beta-interferons, natalizumab, and fingolimod. AQP4-IgG seropositive NMOSD has a relapsing course in most cases, and preventative maintenance treatments should be started after the initial attack. Rituximab, eculizumab, inebilizumab, and satralizumab all have class 1 evidence for use in AQP4-IgG seropositive NMOSD, and the latter three have been approved by the US Food and Drug Administration (FDA). MOGAD is much more likely to be monophasic than AQP4-IgG seropositive NMOSD, and preventative therapy is usually reserved for those who have had a disease relapse. There is a lack of any class 1 evidence for MOGAD preventative treatment. Observational benefit has been suggested from oral immunosuppressants, intravenous immunoglobulin (IVIg), rituximab, and tocilizumab. Randomized placebo-controlled trials are urgently needed in this area.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Aquaporin-4-IgG Seropositive Neuromyelitis Optica Spectrum Disorders (AQP4-IgG Seropositive NMOSD) and Myelin Oligodendrocyte Glycoprotein Antibody–Associated Disorder (MOGAD) — Background
Aquaporin-4-IgG seropositive neuromyelitis optica spectrum disorder (AQP4-IgG seropositive NMOSD) and myelin oligodendrocyte glycoprotein antibody (MOG-IgG)–associated disease (MOGAD) are acquired inflammatory demyelinating diseases of the central nervous system (CNS), distinct from multiple sclerosis (MS). Prior to the discovery of their antibody biomarkers, these diseases were initially often categorized as a subtype of MS. They are now recognized as distinct diseases with important treatment and prognostic differences from each other and from MS.
In 2004 and 2005, an antibody biomarker of NMOSD was discovered that targets aquaporin-4, a water channel on the cell surface of astrocytes [1, 2]. This was the first serum biomarker of any CNS demyelinating disease. AQP4-IgG seropositive NMOSD is relapsing in 90% of cases and attacks are often severe with incomplete recovery and thus disability will accumulate with each attack, further highlighting the importance of attack-prevention treatments in these patients [3, 4]. AQP4-IgG seropositive NMOSD is characterized by three major attack types: (1) transverse myelitis; (2) optic neuritis; (3) area postrema syndrome (which can lead to intractable nausea, vomiting and hiccups from involvement of the brain’s vomiting center) (5). Other attack types include acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome, and symptomatic cerebral syndrome with NMOSD-typical brain lesions [5]. AQP4-IgG seropositive NMOSD affects females 5–10 times more frequently than males and has a higher prevalence in non-white populations [6,7,8]. The median age of disease onset is 35–37 years, but onset has been described in both young children and elderly patients [3, 8, 9]. Unlike MS, it is generally not associated with a secondary progressive course [10]. The latest generation of AQP4-IgG cell-based assays are highly specific (> 99%) for this diagnosis [11,12,13,14,15]. AQP4-IgG seronegative NMOSD cases are described and account for 20% of NMOSD but represent a heterogeneous group of disorders and will not be a major focus of this review [2, 16, 17].
In 2007, when using assays to detect antibodies to the MOG protein in its native conformational form, it was first recognized that MOG-IgG is a biomarker of a distinct CNS demyelinating disease [18]. Using updated cell-based assays, the true spectrum of MOGAD has been elucidated and its distinction from MS has become clearer. The disease is associated most often with attacks of acute disseminated encephalomyelitis (ADEM), optic neuritis, transverse myelitis, cerebral cortical encephalitis, or combinations thereof [19,20,21,22]. It can also account for some patients with the syndrome NMOSD who lack AQP4-IgG. The disease can be monophasic in up to half of patients and the remainder follow a relapsing course. Unlike MS, a secondary progressive course is not encountered [23]. Onset is typically in the third decade of life, with up to half of all cases occurring in children [24,25,26,27]. In contrast with AQP4-IgG seropositive NMOSD, males and females are equally affected, and major differences in race/ethnicity have not been described [27,28,29,30]. Cell-based assays are optimal for detection of MOG-IgG and have high specificity (≈98%) but false positives occur more commonly than with AQP4-IgG, particularly with low positive results (titer 1:20, 1:40 on flow cytometric assay at Mayo Clinic) or when ordered in low probability situations [31, 32]. Cell-based assays can be carried out using live cells or fixed cells (chemically or physically “fixed” to preserve the shape and contents of the cell even though they are dead) [33]. Live cells preserve the membrane integrity and better recapitulate what happens in humans. Moreover, there is concern that fixed cells may allow the antibody to bind to intracellular components of the cell surface receptor or protein of interest which may be less clinically important given antibodies would not have access to such parts of the receptor in real life. Live cell-based assays appear to have slightly higher specificity than fixed cell-based assays [34].
In this article, we will discuss monoclonal antibody (mAb) treatments used in AQP4-IgG seropositive NMOSD and MOGAD, other than mAbs targeting complement. For a detailed discussion of therapies targeting the complement pathway in NMOSD and beyond, we refer the reader to the “anti-complement agents for autoimmune neurological diseases” accompanying article in this issue of neurotherapeutics.
Aquaporin-4-IgG Seropositive Neuromyelitis Optica Spectrum Disorders
Immunopathogenesis
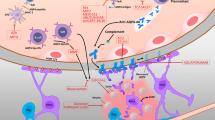
AQP4-IgG seropositive NMOSD is an autoimmune astrocytopathy [35]. AQP4-IgG plays a direct causative role in the pathogenesis of NMOSD [36, 37]. Evidence for the pathogenic mechanism comes from studies involving injection of animals with human AQP4-IgG, from pathology specimens, and from biomarkers in sera and CSF of affected patients. AQP4-IgG is produced by B cells and plasmablasts in the periphery and enters the CNS at areas of increased blood–brain barrier (BBB) permeability or BBB damage. It binds to AQP4 on astrocyte foot processes and causes injury through (1) AQP4 internalization [38, 39], (2) glutamate toxicity via downregulation of excitatory amino acid transporter 2 (EAAT2) [40], (3) generation of reactive astrocytes and activation of the NF-kB pathway [41], (4) complement-dependent cytotoxicity (CDC) [37, 42, 43], and (5) antibody-dependent cellular cytotoxicity (ADCC) [44]. Gaining an understanding of the pathogenic mechanisms has allowed us to develop and use targeted therapies (Fig. 1).

Main pathogenic mechanism by which AQP4-IgG causes NMOSD. 1 — B cells differentiate into AQP4-IgG-secreting plasmablasts which is helped by interleukin-6; 2 — AQP4-IgG enters the circulation and traverses the blood–brain barrier; 3 — AQP4-IgG binds to AQP4 on the surface of astrocytes; 4 — C1q binds to AQP4-IgG and activates the classical complement pathway; 5 — astrocyte damage occurs from opsonization with complement and formation of the membrane attack complex (c5–c9); 6 — C5a is an anaphylatoxin and recruits granulocytes; 7 — granulocytes damage neurons and oligodendrocytes; 8 — the end result is demyelination
B Cells
Naïve B cells, as part of their development, go through the important checkpoints of central and peripheral tolerance, where naïve cells that react to a self-antigen are eliminated. In autoimmune diseases, such as AQP4-IgG seropositive NMOSD, there is a failure of this tolerance process, and autoreactive and polyreactive naïve B cells are able to survive [45, 46].
B cell recruiting and activating factor (BAFF), a proliferation-inducing ligand (APRIL), and C-X-C motif chemokine 13 (CXCL13) are upregulated in the CSF of patients with NMOSD [47,48,49].
A subpopulation of B cells (CD19intCD27highCD38highCD180− B cells) with properties of plasmablasts is thought to be responsible for most of the AQP4-IgG production in NMOSD [50]. Notably, these cells do not express CD20 [50]. There is evidence to suggest that AQP4-IgG-producing plasmablasts enter the CNS from the periphery and establish inflammatory foci [51].
There is also an overall imbalance of the pro-inflammatory and anti-inflammatory B cells in AQP4-IgG seropositive NMOSD [52]. Most of the AQP4-IgG-producing plasma cells originate in the periphery but some intrathecal production has also been described, likely from both novel and peripherally derived B cell clones [53, 54]. There is some evidence that peripherally activated B cells form ectopic tertiary lymphoid structures in the intrathecal compartment, where they undergo clonal expansion and differentiation into plasmablasts [55].
Complement
Most AQP4-IgG is of the IgG1 isotype which is effective at activating the classical complement pathway [39]. This leads to opsonization of astrocytes and their lysis through the formation of the membrane attack complex. Complement activation also causes inflammation through C5a, recruiting other inflammatory cells, such as granulocytes and macrophages, which then cause damage to oligodendrocytes and neurons as well as further compromising BBB integrity [56,57,58,59]. For further information on the role of complement in NMOSD, we refer the reader again to “anti-complement agents for autoimmune neurological diseases” in this issue.
Interleukin 6
Interleukin 6 (IL-6) is a pro-inflammatory cytokine and another key player in the pathogenesis of AQP4-IgG seropositive NMOSD. IL-6 levels are elevated in patients with NMOSD, particularly during attacks [60,61,62,63,64]. AQP4-IgG binding promotes IL-6 release from astrocytes [65]. IL-6 contributes to the pathology of NMOSD by (1) effect on B cells, promoting differentiation and maintenance of plasmablasts from B cells and enhancing secretion of AQP4-IgG [50, 66]; (2) effect on T cells, promoting the differentiation and maintenance of naive T cells to proinflammatory T-helper-17 cells and inhibiting activation of regulatory T cells [67,68,69]; (3) effect on innate immune system, activates innate immune cells [70]; and (4) effect on BBB, increased permeability [65].
Therapeutic Strategies
Treatment of AQP4-IgG seropositive NMOSD can be divided into acute treatments of attacks and chronic maintenance attack-prevention treatments, the latter of which will be the focus of this review.
Acute Treatment of Attacks
Treatment of acute attacks consists of high-dose IV steroids and plasma exchange. Given the attacks are often severe with incomplete recovery, we have a very low threshold for giving plasma exchange in addition to corticosteroids. Indeed, retrospective studies have suggested evidence for the early use of plasma exchange in improving outcomes [71, 72]. Dosing of attack-prevention mAb treatments should be undertaken after plasma exchange has been completed, to avoid plasma exchange removing the attack-prevention monoclonal antibody.
Bevacizumab is a mAb which has undergone some studies to assess its potential utility in the acute treatment of AQP4-IgG seropositive NMOSD, although is not yet part of the standard treatment regimen. Bevacizumab is a humanized mAb that targets vascular endothelial growth factor A (VEGF-A), inhibiting the formation of new blood vessels [73]. The rationale for the use of bevacizumab in AQP4-IgG seropositive NMOSD is that astrocyte-derived VEGF-A drives BBB disruption in CNS inflammatory disease [74]. In 2015, a single-center, open-label phase 1b trial for bevacizumab in acute treatment of attacks was published [75]. It was a safety and proof of concept study involving 10 patients with AQP4-IgG seropositive NMOSD. Bevacizumab (10 mg/kg intravenously) was administered on day 1, in addition to a 5-day course of methylprednisolone (1 g intravenously). Three patients recovered to pre-attack neurological function or better, and no patients required escalation to plasmapheresis. Bevacizumab was safe in all 10 participants, with only one serious adverse event occurring within the 90-day follow-up, and this was not attributed to the medication. Larger randomized control trials are needed to properly assess the efficacy and safety of bevacizumab prior to it being incorporated into clinical practice.
Chronic Treatment to Prevent Attacks
In general, it is recommended that all patients with AQP4-IgG seropositive NMOSD be started on attack prevention treatments from onset given the potential severity of attacks and risk of incomplete recovery (a single attack can result in permanent blindness, paraplegia, or death from neurogenic respiratory failure) [76]. Initial studies showed that some MS treatments, such as IFN-β, natalizumab, alemtuzumab, and fingolimod can worsen AQP4-IgG seropositive NMOSD [77,78,79,80]. Early treatments used for AQP4-IgG seropositive NMOSD were focused on broad oral immunosuppressants utilized in the treatment of other autoimmune neurologic disorders and retrospective studies reported some possible benefit of mycophenolate and azathioprine [81]. However, these treatments take a long time to take effect and thus require concomitant corticosteroids for 3–6 months which can have a high side effect burden. With increasing knowledge of the immunopathogenesis of AQP4-IgG seropositive NMOSD, targeted mAb treatments to prevent attacks were developed and have been studied in clinical trials. In the following sections, we will discuss novel mAb treatments used in AQP4-IgG seropositive NMOSD other than monoclonal antibodies targeting complement. For a detailed discussion of therapies targeting the complement pathway in NMOSD and beyond, we refer the reader to the “anti-complement agents for autoimmune neurological diseases” accompanying article in this issue of neurotherapeutics. It is of note that transitional oral corticosteroids at low doses (20–30 mg oral prednisone daily followed by a taper) are often used to reduce the risk of early attacks while the prevention treatments take effect, which usually takes just a few weeks with these novel mAb treatments.
There are four mAbs with class 1 evidence for use (rituximab, inebilizumab, satralizumab, and eculizumab) [82], three of which have been approved by the US Food and Drug Administration (FDA) for the treatment of AQP4-IgG seropositive NMOSD (inebilizumab, eculizumab, and satralizumab) (Table 1). These treatments are very effective and although also very expensive, their use likely reduces downstream costs that would be incurred from breakthrough attacks which often require prolonged hospitalization and expensive attack treatment [83].
Treatment Options
Anti-CD20 Therapies
Rituximab
Therapeutic Mechanism
Rituximab is a chimeric monoclonal IgG1 that binds to CD20 on B cells. Rituximab depletes CD20 + B cells in peripheral blood, with one goal to try to reduce the levels of AQP4-IgG. Other mechanisms of action may include inhibition of B/T-cell interactions, decreasing pro-inflammatory cytokines, and modulating the T-cell compartment [84,85,86]. BAFF is a B cell survival factor which supports autoreactive B cells and prevents their deletion [87]. Rituximab increases BAFF levels [88]. Belimumab, a BAFF inhibitor that has shown efficacy when used after rituximab in patients with systemic lupus erythematosus, may also be a consideration for investigation for NMOSD [89].
Evidence
Prior to the evidence from randomized controlled trials (RCTs) for newer treatments, rituximab was commonly used and recommended as first-line maintenance therapy for AQP4-IgG seropositive NMOSD, given its efficacy in other autoimmune diseases [90, 91]. Rituximab showed efficacy for relapse prevention in multiple case series and retrospective analyses [92,93,94,95,96,97,98]. An open-label trial showed benefit of rituximab over azathioprine for relapse prevention [99].
RIN-1
Trial Design
The RIN-1 study is a multicenter, randomized, double-blind, placebo-controlled clinical trial performed at eight hospitals in Japan, and published in 2020 [100]. Only AQP4-IgG seropositive patients were included. Patients were matched 1:1 for rituximab (intravenous treatment with 375 mg/m2 every week for 4 weeks followed by two 1000 mg infusions 2 weeks apart at six monthly intervals) and placebo. Both groups had 19 patients. Relapse had occurred during the 2 years prior to enrollment in 12/19 (63%) patients assigned to rituximab and 11/19 (58%) assigned to placebo. The study period was 72 weeks.
Efficacy
None (0/19 [0%]) of the patients on rituximab had relapses during the study period, compared to 7/19 (37%) of patients on placebo (CI: 12.3–65.5%; p value = 0.0058). However, the small sample size did not allow a reliable calculation of risk reduction by rituximab treatment. A post hoc analysis of AQPR-IgG titers found that in patients assigned placebo, titers increased or remained high and did not decrease in any patients. By contrast, in patients assigned rituximab, AQP4-IgG titers decreased gradually in six (32%) patients, suggesting that rituximab prevented relapses without reducing AQP4 antibody titers in the remaining 13 patients.
Adverse Events
Adverse events were reported in 17/19 (90%) patients in both groups. Infusion reactions were more common in the rituximab group. No deaths occurred.
Ublituximab
Therapeutic Mechanism
Ublituximab is a chimeric IgG1 targeting CD20 with a low fucose content of oligosaccharides [101]. The low fucose content allows it to bind with higher affinity to FcγRIIIa, which increases its ADCC activity to 100 times more than rituximab [102,103,104].
Evidence
In 2019, results from an open-label phase 1 study were published. This was a safety and proof of concept trial in five patients with AQP4-IgG seropositive NMOSD [105]. Ublituximab (450 mg intravenously) was administered once, within 5 days of relapse onset, in addition to methylprednisolone (1 g intravenously for 5 days).
The primary outcome was safety with secondary efficacy measures. Expanded Disability Status Scale (EDSS) scores dropped from admission (median = 6.5) to 90-day follow-up (median = 4). Two subjects did not achieve total B cell depletion and relapsed within 3 months.
Although the adverse events of ublituximab were mostly immunosuppressive, there were no opportunistic infections or serious adverse events. Common side effects included diarrhea, constipation, fatigue, and neutropenia.
Anti-CD19 Therapies
Inebilizumab
Therapeutic Mechanism
Inebilizumab is a humanized IgG1 targeting CD19, which has broader expression than CD20. CD20 is present on pre–B cells; naïve, mature, memory B cells; and some plasmablasts [106], whereas CD19 is a pan B cell marker, also present on pro-B cells, plasmablasts, and some plasma cells [107]. CD19 is also more selective for B cells, while CD20 can also be expressed on some T cells [52, 85, 108, 109]. Hence, CD19 might be a more attractive target than CD20 for B cell–directed therapies in AQP4-IgG seropositive NMOSD.
Evidence
N-MOmentum
Trial Design
N-MOmentum is a multicenter, double-blind, randomized placebo-controlled, phase 2/3 trial for inebilizumab published in 2019 [110]. It was carried out across 99 centers in 25 countries. Patients were randomized 3:1 to the inebilizumab (n = 174) and placebo (n = 56) groups. Inebilizumab was administered intravenously at a dose of 300 mg (initial loading of 2 doses 2 weeks apart and then 6 monthly). The primary endpoint was time to onset of an NMOSD relapse.
Efficacy
The trial was terminated early by the independent data-monitoring committee, due to a clear demonstration of efficacy. Only 21/174 (12%) of patients in the inebilizumab group had an attack of NMOSD, compared to 22/56 (39%) in the placebo group (hazard ratio 0.272 [95% CI 0.15–0.496]; p < 0.0001). The efficacy was even better for AQP4-IgG seropositive patients, with relapse occurring in 18/161 (11%) in the inebilizumab group vs 22/52 (42%) in the placebo group (p < 0.001). Although prior studies suggest that titers do not impact relapses [111, 112], AQP4-IgG titers after treatment have not yet been analyzed and may offer insights into the mechanism of action of inebilizumab.
Adverse Events
Both groups had similar adverse event rates (72% inebilizumab vs 73% placebo). Serious adverse events were reported more frequently in the placebo group (9% vs 5%). One death occurred in each group. The cause of death for the patient in the placebo group was respiratory insufficiency related to NMOSD relapse. The cause of death for the patient in the inebilizumab arm was a CNS process of unclear etiology (the differential diagnoses were acute disseminated encephalomyelitis, atypical NMOSD attack, and progressive multifocal leukoencephalopathy).
Additional Analyses
A post hoc analysis of 75 AQP4-IgG seropositive patients receiving inebilizumab for ≥ 4 years (randomized controlled period and open-label extension of the N-MOmentum study) found that 18 attacks occurred in 13 patients [113]. Of these, 12 attacks occurred in the first year of treatment and two per year in years 2–4. This suggests that the efficacy of inebilizumab may be enhanced after the first year of treatment. This may apply to all B cell–targeted treatments as early relapse after treatment has also been described with rituximab [114]. Reasons for this phenomenon are unclear but potentially include the time taken for the autoantibody to be removed from circulation, an increase in pro-inflammatory cytokines such as BAFF, and the release of AQP4-IgG from B cells [114]. Further studies assessing B cell depletion, AQP4-IgG titers, and BAFF levels, over time with inebilizumab treatment, are warranted to answer this question. Inebilizumab was well tolerated, with two serious adverse events related to inebilizumab in the study period and no deaths. IgG levels decreased over time; however, correlation between severe infections and low IgG levels could not be determined because of the small numbers [113]. A subgroup analysis found that the benefit of inebilizumab vs placebo remained, regardless of attack definition, type of attack, baseline disability, ethnicity, treatment history, or disease course [115]. Another analysis showed that compared with placebo, inebilizumab reduced the risk of 3-month EDSS-confirmed disability progression [116].
Infection Risk in CD19 and CD20 Targeted Treatments
Anti-B cell therapies have the potential to cause hypogammaglobulinemia. Although unproven, the risk theoretically may be greater with anti-CD19 than anti-CD20 treatments, given that CD19 is expressed on a wider repertoire of B cells. In the N-MOmentum trial, at day 197, IgG levels had decreased by 4.0% in the inebilizumab group and increased by 6.2% in the placebo group [110]. Hypogammaglobulinemia can lead to secondary antibody deficiency (SAD) with an increase in bacterial infections of the sinopulmonary and gastrointestinal tract and of viral infections such as enterovirus. Hypogammaglobulinemia can be transient and potentially reversible (partially or fully) but has the potential to limit the long-term use of B cell–depleting therapies [117, 118]. A study of ocrelizumab (anti-CD20) in patients with MS found that 5.4% of patients had low IgG levels after 5 years of treatment [119]. Lower baseline immunoglobulin levels result in lower post treatment levels and autoimmunity can occasionally be the first manifestation of primary immunodeficiency [120,121,122]. We therefore recommend checking pre-treatment immunoglobulin levels, in addition to hepatitis B/C and HIV serology. The role of testing for latent tuberculosis (TB) is less clear. While some physicians only do QuantiFERON testing in high-risk patients, the American College of Rheumatology recommends a more cautious approach with testing for all patients prior to rituximab treatment [123, 124]. A study in a TB endemic area of the UK showed a 10% prevalence of latent TB in patients being treated with biologic therapy [125]. Even in non-endemic areas, latent TB infection can be frequent (> 1%) in patients receiving immunotherapy, which would support the need for screening of all patients [126]. We also recommend three monthly CBC and yearly IgG monitoring [127]. If IgG levels are low (especially < 500 mg/dL and/or recurrent infections), consider clinical immunology consult who can advise on the need for prophylactic antimicrobials and/or replacement with IVIg (0.4 g/kg once a month initially, titrated to effect on infection rate and/or to achieve a minimum IgG concentration of 800 mg/dL) [127]. The number of patients requiring immunoglobulin replacement following rituximab was 4.5% in a study of 8663 patients [128]. Note that the replacement dose is much lower than the immunomodulatory dose of IVIg used for the treatment of immune-mediated disorders. Hypogammaglobulinemia (pre- or post treatment) is not a contraindication to further B cell–targeted therapies [118, 129].
Anti-interleukin 6 Therapies
Tocilizumab
Therapeutic Mechanism
Tocilizumab is a humanized mAb targeting the IL-6 receptor.
Evidence
Several retrospective case series and reports suggested efficacy of tocilizumab in the treatment of NMOSD [130,131,132,133].
Three studies showed that tocilizumab led to an absolute risk reduction (ARR) for NMOSD relapses [130, 134, 135]. Two of the studies were in patients who had failed B cell therapies.
TANGO
Trial Design
The TANGO trial, published in 2020, is an open-label, multicenter, randomized, phase 2 trial for tocilizumab in NMOSD [136]. Patients were randomized 1:1 to receive treatment with tocilizumab (8 mg/kg every 4 weeks intravenously) or azathioprine (2–3 mg/kg/day orally), with 59 patients in each group. In total, 85% of patients in the tocilizumab group and 90% of patients in the azathioprine group were AQP4-IgG seropositive. The study period was 60 weeks.
Efficacy
Relapse rates were lower in the tocilizumab group (8/59, 14%) than the azathioprine group (28/59, 47%) (hazard ratio [HR] 0.236 [95% CI 0.107–0.518]; p < 0.0001).
Adverse Events
Tocilizumab had a lower rate of treatment-associated adverse events than azathioprine (61% vs 83%). The most commonly reported adverse events were increased alanine transaminase concentrations, upper respiratory tract infections, and urinary tract infections. There was one death in each group. The cause of death in the tocilizumab group was myelitis ascending to the medulla oblongata. In the azathioprine group, the cause of death was acute meningoencephalitis caused by Listeria monocytogenes.
Satralizumab
Therapeutic Mechanism
Satralizumab is another humanized mAb targeting the IL-6 receptor. It was designed using recycling antibody technology™ to last longer in the circulation, and thus, have a longer half-life than tocilizumab [137, 138].
Evidence
SAkuraSky
Trial Design
This was an international, randomized, double-blind, placebo-controlled, phase 3 trial [139]. It included AQP4-IgG seropositive and seronegative NMOSD patients. Patients received satralizumab (n = 41) or placebo (n = 42) in addition to their baseline oral immunosuppressive therapy. Satralizumab dosing was 120 mg subcutaneously at 0, 2, and 4 weeks and every 4 weeks thereafter. The median treatment duration with satralizumab was 107.4 weeks.
Efficacy
Patients in the satralizumab treatment arm had a lower rate of relapse (8/41, 20%) than the placebo arm (18/42, 43%) (hazard ratio, 0.38; 95% confidence interval [CI], 0.16 to 0.88). Subgroup analysis showed better efficacy in patients with AQP4-IgG seropositive NMOSD (relapse occurred in 11% (3/27) of those in the satralizumab group and in 43% (12/28) of those in the placebo group (hazard ratio, 0.21; 95% CI, 0.06 to 0.75)). No differences were found in the AQP4 seronegative subgroup subanalysis.
Adverse Events
The rates of serious adverse events (satralizumab 17%, placebo 21%) and infections (satralizumab 68%, placebo 62%) were similar in both groups. Injection site reactions were more frequently reported by patients in the satralizumab group (12% vs 5%). Other common adverse effects included neutropenia and elevated liver enzymes. No deaths occurred.
SAkuraStar
Trial Design
This was an international, randomized, double-blind, placebo-controlled, phase 3 trial [140]. It was carried out across 44 centers in 13 countries and included AQP4-IgG seropositive and seronegative NMOSD patients. In contrast to SAkuraSky, satralizumab was administered as monotherapy and taking other immunosuppressants concomitantly was prohibited. Patients were randomly assigned to receive satralizumab (n = 63) or placebo (n = 32). Treatment duration was until 44 protocol-defined relapses occurred or 1.5 years after random assignment of the last patient enrolled, whichever occurred first.
Efficacy
Relapse occurred in 19/63 (30%) of patients in the satralizumab arm and 16/32 (50%) in the placebo arm (hazard ratio 0.45, 95% CI 0.23–0.89; p = 0.018). For AQP4-IgG seropositive patients, relapse occurred in 9/41 (22%) in the satralizumab group versus 13/23 (57%) in the placebo group (95% CI, 0.11–0.63).
Adverse Events
Both groups had similar rates of serious adverse events (19% satralizumab vs 16% placebo) and infections (54% satralizumab vs 44% placebo). Patients on satralizumab did not report a higher frequency of injection site reactions in this trial (13% vs 16%).
Monitoring Requirements for Anti-IL-6 Therapies
Due to the effect on liver transaminases, monitoring of liver function tests is recommended (every 4 weeks for the first 3 months of treatment, every 3 months for 1 year, annually thereafter) in addition to CBC monitoring for neutropenia (4–8 weeks after initiation of therapy and annually thereafter) [127].
Emerging Treatments
Aquaporumab
Aquaporumab is a non-pathogenic human mAb that binds to AQP4 with high affinity. It was generated from a recombinant pathogenic monoclonal AQP4-IgG [141, 142]. The Fc (fragment crystallizable) region of aquaporumab has been artificially mutated to prevent its CDC and ADCC effects. It competitively displaces AQP4-IgG from AQP4. Aquaporumab reduced NMO lesions in vitro models and in mice [143]. Given its highly specific mechanism of action, minimal side effects of treatment are anticipated but could include formation of antiidiotypic host antibodies against aquaporumab. Aquaporumab looks to be a promising treatment; however, a decade after its creation, there is no start date for clinical trials [144]. Moreover, we do not know of targeting a specific variable region epitope or epitopes is enough to block the polyclonal anti-AQP4 response and do not know for sure if it could be immunogenic itself. All of this will need to be addressed in human studies.
MOG-IgG-Associated Disease (MOGAD)
Pathogenesis
MOG is expressed on the surface of oligodendrocytes and is a minor component of the myelin sheath. The function of MOG has not been fully elucidated but evidence suggests it plays a role in adhesion of myelin fibers, regulation of microtubule stability, and myelin-immune system interactions via the complement pathway [145,146,147]. The majority of human MOG antibodies do not recognize rodent MOG, which has hampered applying animal MOG-IgG studies to human MOGAD. However, studies that used only human MOG-IgG that does recognize rodent MOG, and injected them intrathecally along with myelin reactive T-cells, found that this resulted in demyelination and complement deposition, suggesting pathogenic potential of the antibody [148]. This study used high titer antibodies and thus could suggest that higher titers are more likely to be pathogenic, whereas prior studies have shown that low titer results may be found in healthy and disease controls [30, 148, 149]. Though there is some evidence that persistently positive MOG-IgG titers are predictive of relapse, further studies, to evaluate the impact of seroconversion, are needed before MOG-IgG titers can be used as a marker to assess therapeutic efficacy [22, 150, 151]. It will be important to include MOG-IgG titers as a surrogate biomarker and correlate the values with outcome in upcoming clinical trials of MOGAD in order to determine if it could be a marker of therapeutic efficacy. The location of MOG-IgG production is yet to be fully elucidated though two sources of circulating autoantibodies have been proposed: plasmablasts that emerge from germinal center reactions in secondary lymphoid organs and long-lived plasma cells located in the bone marrow [55]. B cells activated in secondary lymphoid tissue may also migrate into the intrathecal compartment to undergo clonal expansion and differentiate into plasmablasts, potentially associated with the formation of ectopic tertiary lymphoid structures [152]. Recent studies have shown higher CSF MOG-IgG levels than expected from the patient’s serum level as well as occasional circumstances when MOG-IgG is detected in the CSF and not serum, which is also suggestive of intrathecal synthesis of MOG-IgG [153, 154]. MOG-IgGs from most patients require bivalent binding. Since bivalently bound antibodies have been reported to only poorly bind C1q, the complement pathway may play a less active role in MOGAD than AQP4-IgG seropositive NMOSD [155]. However, the role of complement in MOGAD is controversial and remains yet to be fully determined. Specimens from biopsy and autopsy reveal perivenous as well as confluent cortical and white matter demyelination, CD4 + T cell and granulocyte infiltration, and complement deposition, and some studies have suggested loss of MOG immunostaining and others have not [156,157,158]. Unlike AQP4-IgG seropositive NMOSD, with MOGAD, the AQP4 immunostaining is preserved and astrocytes are also typically preserved [156,157,158]. In contrast to MS in which MRI T2-lesions persist over time, MOGAD T2-lesions often resolve completely in follow-up which might provide some insight into the better long-term prognosis and absence of a secondary progressive course in MOGAD [159]. The pathophysiologic underpinning of this much higher frequency of lesion resolution when compared to MS is uncertain but it could relate to enhanced ability to remyelinate in MOGAD [159].
B Cells
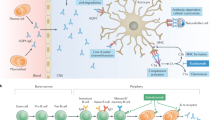
In comparison to healthy controls, patients with MOGAD (similar to AQP4-IgG seropositive NMOSD) may have an imbalance in proinflammatory and anti-inflammatory B cells, with decreased regulatory and IL-10 + B cells and increased memory B cells as well as increased T-follicular helper cels (Tfh) [160]. However, it is important to remember that these results were from an immunophenoty** study with intracellular staining. Immunophenoty** studies are rarely reproduced and only allow for limited conclusions about the function of the cells studied. Naïve B cells are activated by Tfh cells, the activated B cells then differentiate to MOG-IgG-secreting plasmablasts (Fig. 2). Less than 50% of MOGAD patients harbor MOG-specific B cells in the peripheral blood, which are not linked to levels of MOG-IgG in serum, suggesting different sources of MOG-IgG [161]. Identification of MOG-specific B cells in blood could be of future relevance for selecting patients with MOG-IgG for B cell–directed therapy [161].

Main pathogenic mechanism of MOGAD. 1 — B cells differentiate into MOG-IgG-secreting plasmablasts which is helped by interleukin-6; 2 — MOG-IgG enters the circulation and traverses the blood–brain barrier; 3 — there is also some intrathecal production of MOG-IgG; 4 — MOG-IgG binds to MOG protein on the surface of oligodendrocytes; 5 — inflammation occurs with release of cytokines such as interleukin-6 and BAFF and CD4 + T cells and macrophages are recruited; 6 — these cells damage neurons and oligodendrocytes; the end result is demyelination
Interleukin-6
There are raised levels of Th17-related cytokines, such as IL-6, in the CSF of MOGAD patients during an attack. As described above, this is also seen in AQP4-IgG seropositive NMOSD but not in MS [162].
Therapeutic Strategies
Treatment of MOGAD can also be divided into acute treatments of attacks and chronic maintenance attack prevention treatments, again we will focus on the latter.
Acute Treatment of Attacks
We recommend acute treatment of all attacks. There is evidence to suggest that early glucocorticoid treatment could prevent residual damage [163]. First-line treatment is with 1 g of IV methylprednisolone daily for 5 days, with high-dose oral prednisolone 1250 mg daily for 5 days as an alternative. An oral steroid taper over weeks to months is sometimes considered to prevent early recurrence on steroid withdrawal. Most patients respond well to glucocorticoids [30]. For those that do not, second-line options are plasma exchange and IVIg (particularly in children) [164].
Chronic Treatment to Prevent Attacks
MOGAD is more likely to have a monophasic course with better recovery from attacks, when compared to AQP4-IgG seropositive NMOSD [151]. Due to the high proportion of people whose disease will remain monophasic (40–50%), preventative treatment is usually commenced only after a relapse occurs. There are exceptions to this rule, particularly if the first attack was life threatening or leaves the patient with severe residuals deficits (e.g., unilateral blindness). Similar to AQP4-IgG seropositive NMOSD, certain disease-modifying drugs used in MS (IFN-β, fingolimod, and natalizumab) do not appear to be effective [80, 165, 166]. Treatment options include oral immunosuppression, IVIg, and mAbs. In a study assessing the efficacy of different immunosuppressive therapies for relapse prevention in MOGAD, IVIg appeared to have the lowest annualized relapse rate, followed by azathioprine, then rituximab [166]. Evidence for treatment comes from case series and case reports, mostly retrospective. With no class 1 evidence and no FDA-approved treatments, prospective, placebo-controlled randomized controlled trials are urgently needed.
Treatment Options
Anti-CD20
Rituximab
Several studies, including a meta-analysis, have shown a reduced risk of relapse with rituximab. However, it appears to be less effective than for AQP4-IgG seropositive NMOSD with relapse occurring in up to half of all patients, sometimes despite B cell depletion [167].
Anti-IL-6 Receptor
Tocilizumab
Tocilizumab reduced the risk of MAGAD relapse in retrospective case series [133, 168].
Emerging Treatments
Rozanolixizumab
Rozanolixizumab is a humanized IgG4 mAb targeting the neonatal Fc receptor (FcRn). FcRn is responsible for IgG recycling intracellularly and inhibiting it leads to accelerated elimination of IgG. Rozanolixizumab hence reduces plasma IgG levels [169, 170]. Phase 2 trials have shown its efficacy and safety in myasthenia gravis, and clinical trials for MOGAD are due to begin. Headaches are the most common side effect, occurring in up to 39% of patients [171, 172].
Discussion
There have been no head-to-head trials to compare the different monoclonal treatment options for NMOSD and MOGAD. Direct comparison is difficult due to differences in trial design and baseline patient characteristics. For example, when comparing B cell–depleting therapies for NMOSD, patients in the treatment group in RIN-1 had no relapses, whereas 12% of those in the treatment group in N-MOmentum had a breakthrough relapse. However, RIN-1 had a much smaller number of patients and patients had lower relapse rates in the 2 years prior to recruitment so there is no sufficient evidence to conclude that rituximab is a superior treatment to inebilizumab. By understanding the pathophysiology of these diseases, we have been able to develop highly targeted, effective treatments. In turn, the efficacy of these target treatments contributes to our understanding of the pathophysiological mechanisms. In the absence of definitive evidence of superiority of one drug, other factors that may influence treatment choice include whether the treatment is licensed, physician experience/comfort with the treatment, adverse effect profile, accessibility, patient preference, and cost.
Conclusions
Disease-modifying treatments used in MS are not effective for AQP4-IgG seropositive NMOSD or MOGAD. Acute treatment of NMOSD is with high-dose IV steroids and plasma exchange. AQP4-IgG seropositive NMOSD has four attack-prevention treatment options with class 1 evidence for use: eculizumab, inebilizumab, rituximab, and satralizumab. Tocilizumab also has some evidence for use and aquaporumab is a promising emerging therapy. Treatment is initiated after the first attack. Acute treatment of MOGAD is with corticosteroids and if refractory to steroids, plasma exchange or IVIg. There have been no randomized controlled trials for attack-prevention treatment of MOGAD, with observational evidence for oral immunosuppressants, IVIg, anti-B cell, and anti-IL-6 therapies. Since MOGAD can often have a monophasic course, treatment is often not started unless the patient has a relapse of disease.
References
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106–12.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–7.
Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(9):1176–80.
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107–14.
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177–89.
Wingerchuk DM. Neuromyelitis optica: effect of gender. J Neurol Sci. 2009;286(1–2):18–23.
Papp V, Magyari M, Aktas O, Berger T, Broadley SA, Cabre P, et al. Worldwide incidence and prevalence of neuromyelitis optica: a systematic review. Neurology. 2021;96(2):59–77.
Flanagan EP, Cabre P, Weinshenker BG, Sauver JS, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79(5):775–83.
Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70(5):344–52.
Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603–5.
Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neurol. 2014;5(3):290–303.
Fryer J, Lennon V, Pittock S, Jenkins S, Fallier-Becker P, Clardy S, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm. 2014;1(1).
Prain K, Woodhall M, Vincent A, Ramanathan S, Barnett MH, Bundell CS, et al. AQP4 Antibody assay sensitivity comparison in the era of the 2015 Diagnostic Criteria for NMOSD. Front Neurol. 2019;10:1028.
Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2016;87(9):1005–15.
Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol. 2013;23(6):661–83.
Akman-Demir G, Tüzün E, Waters P, Içöz S, Kürtüncü M, Jarius S, et al. Prognostic implications of aquaporin-4 antibody status in neuromyelitis optica patients. J Neurol. 2011;258(3):464–70.
Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81(14):1197–204.
O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13(2):211–7.
Weber MS, Derfuss T, Brück W. Anti-myelin oligodendrocyte glycoprotein antibody-associated central nervous system demyelination-a novel disease entity? JAMA Neurol. 2018;75(8):909–10.
de Seze J. MOG-antibody neuromyelitis optica spectrum disorder: is it a separate disease? Brain. 2017;140(12):3072–5.
Kunchok A, Chen JJ, Saadeh RS, Wingerchuk DM, Weinshenker BG, Flanagan EP, et al. Application of 2015 seronegative neuromyelitis optica spectrum disorder diagnostic criteria for patients with myelin oligodendrocyte glycoprotein IgG-associated disorders. JAMA Neurol. 2020;77(12):1572–5.
López-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. 2018;75(11):1355–63.
Lopez-Chiriboga AS, Sechi E, Buciuc M, Chen JJ, Pittock SJ, Lucchinetti CF, et al. Long-term outcomes in patients with myelin oligodendrocyte glycoprotein immunoglobulin G-associated disorder. JAMA Neurol. 2020;77(12):1575–7.
Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128–38.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89(2):127–37.
Cobo-Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical Features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. 2021;89(1):30–41.
de Mol CL, Wong Y, van Pelt ED, Wokke B, Siepman T, Neuteboom RF, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler. 2020;26(7):806–14.
O’Connell K, Hamilton-Shield A, Woodhall M, Messina S, Mariano R, Waters P, et al. Prevalence and incidence of neuromyelitis optica spectrum disorder, aquaporin-4 antibody-positive NMOSD and MOG antibody-positive disease in Oxfordshire. UK J Neurol Neurosurg Psychiatry. 2020;91(10):1126–8.
Hyun JW, Lee HL, Jeong WK, Lee HJ, Shin JH, Min JH, et al. Comparison of MOG and AQP4 antibody seroprevalence in Korean adults with inflammatory demyelinating CNS diseases. Mult Scler. 2021;27(6):964–7.
Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20(9):762–72.
Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. 2018;15(1):134.
Sechi E, Buciuc M, Pittock SJ, Chen JJ, Fryer JP, Jenkins SM, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. 2021.
Sampedro-Carrillo EA. Sample preparation and fixation for histology and pathology. Methods Mol Biol. 2022;2422:33–45.
Waters PJ, Komorowski L, Woodhall M, Lederer S, Majed M, Fryer J, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. 2019;92(11):e1250–5.
Fujihara K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol. 2019;32(3):385–94.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6(9):805–15.
Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66(5):630–43.
Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci U S A. 2012;109(4):1245–50.
Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69(24):2221–31.
Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med. 2008;205(11):2473–81.
Howe CL, Kaptzan T, Magaña SM, Ayers-Ringler JR, LaFrance-Corey RG, Lucchinetti CF. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia. 2014;62(5):692–708.
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133(Pt 2):349–61.
Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125(Pt 7):1450–61.
Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, et al. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181(8):5730–7.
von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol. 2010;11(1):14–20.
Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, et al. Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain. 2019;142(6):1598–615.
Wang H, Wang K, Zhong X, Qiu W, Dai Y, Wu A, et al. Cerebrospinal fluid BAFF and APRIL levels in neuromyelitis optica and multiple sclerosis patients during relapse. J Clin Immunol. 2012;32(5):1007–11.
Quan C, Yu H, Qiao J, **ao B, Zhao G, Wu Z, et al. Impaired regulatory function and enhanced intrathecal activation of B cells in neuromyelitis optica: distinct from multiple sclerosis. Mult Scler. 2013;19(3):289–98.
Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. 2014;176(2):149–64.
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108(9):3701–6.
Chihara N, Aranami T, Oki S, Matsuoka T, Nakamura M, Kishida H, et al. Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS One. 2013;8(12):e83036.
Bennett JL, O'Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e104.
Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617–29.
Kowarik MC, Dzieciatkowska M, Wemlinger S, Ritchie AM, Hemmer B, Owens GP, et al. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19.
Sun B, Ramberger M, O’Connor KC, Bashford-Rogers RJM, Irani SR. The B cell immunobiology that underlies CNS autoantibody-mediated diseases. Nat Rev Neurol. 2020;16(9):481–92.
Jasiak-Zatonska M, Kalinowska-Lyszczarz A, Michalak S, Kozubski W. The immunology of neuromyelitis optica-current knowledge, clinical implications, controversies and future perspectives. Int J Mol Sci. 2016;17(3):273.
Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663–79.
Frampton JE. Eculizumab: a review in neuromyelitis optica spectrum disorder. Drugs. 2020;80(7):719–27.
Kuroda H, Fujihara K, Takano R, Takai Y, Takahashi T, Misu T, et al. Increase of complement fragment C5a in cerebrospinal fluid during exacerbation of neuromyelitis optica. J Neuroimmunol. 2013;254(1–2):178–82.
Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Matsuse D, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One. 2013;8(4):e61835.
Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler. 2010;16(12):1443–52.
Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, et al. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2020;7(5).
Wang H, Wang K, Zhong X, Dai Y, Qiu W, Wu A, et al. Notable increased cerebrospinal fluid levels of soluble interleukin-6 receptors in neuromyelitis optica. NeuroImmunoModulation. 2012;19(5):304–8.
Içöz S, Tüzün E, Kürtüncü M, Durmuş H, Mutlu M, Eraksoy M, et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010;120(1):71–5.
Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, et al. Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro. Neurol Neuroimmunol Neuroinflamm. 2017;4(1):e311.
Wilson R, Makuch M, Kienzler AK, Varley J, Taylor J, Woodhall M, et al. Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain. 2018;141(4):1063–74.
Lin J, Li X, **a J. Th17 cells in neuromyelitis optica spectrum disorder: a review. Int J Neurosci. 2016;126(12):1051–60.
Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40(7):1830–5.
Nicolas P, Ruiz A, Cobo-Calvo A, Fiard G, Giraudon P, Vukusic S, et al. The balance in T follicular helper cell subsets is altered in neuromyelitis optica spectrum disorder patients and restored by rituximab. Front Immunol. 2019;10:2686.
Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–8.
Bonnan M, Valentino R, Debeugny S, Merle H, Fergé JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89(4):346–51.
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke K-D, Hellwig K, et al. Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurology(R) neuroimmunology & neuroinflammation. 2018;5(6):e504-e.
Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7(4):335–45.
Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122(7):2454–68.
Mealy MA, Shin K, John G, Levy M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin Exp Neuroimmunol. 2015;6(4):413–8.
Zhao-Fleming HH, Valencia Sanchez C, Sechi E, Inbarasu J, Wijdicks EF, Pittock SJ, et al. CNS demyelinating attacks requiring ventilatory support with myelin oligodendrocyte glycoprotein or aquaporin-4 antibodies. Neurology. 2021;97(13):e1351–8.
Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69(2):239–45.
Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67(8):1016–7.
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18(1):113–5.
Azzopardi L, Cox AL, McCarthy CL, Jones JL, Coles AJ. Alemtuzumab use in neuromyelitis optica spectrum disorders: a brief case series. J Neurol. 2016;263(1):25–9.
Huang W, Wang L, Zhang B, Zhou L, Zhang T, Quan C. Effectiveness and tolerability of immunosuppressants and monoclonal antibodies in preventive treatment of neuromyelitis optica spectrum disorders: a systematic review and network meta-analysis. Mult Scler Relat Disord. 2019;35:246–52.
Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(7):614–25.
Neuromyelitis optica: new therapies offer hope: Mayo Clinic Medical Professionals/ Neurology and neurosurgery; 6/19/2021 [Available from: https://www.mayoclinic.org/medical-professionals/neurology-neurosurgery/news/neuromyelitis-optica-new-therapies-offer-hope/mac-20515747.
Graves J, Vinayagasundaram U, Mowry EM, Matthews IR, Marino JA, Cheng J, et al. Effects of rituximab on lymphocytes in multiple sclerosis and neuromyelitis optica. Mult Scler Relat Disord. 2014;3(2):244–52.
Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4(10):557–67.
Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–40.
Möckel T, Basta F, Weinmann-Menke J, Schwarting A. B cell activating factor (BAFF): structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun Rev. 2021;20(2):102736.
Vallerskog T, Heimbürger M, Gunnarsson I, Zhou W, Wahren-Herlenius M, Trollmo C, et al. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2006;8(6):R167.
Shipa M, Embleton-Thirsk A, Parvaz M, Santos LR, Muller P, Chowdhury K, et al. Effectiveness of belimumab after rituximab in systemic lupus erythematosus: a randomized controlled trial. Ann Intern Med. 2021;174(12):1647–57.
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019–32.
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. 2014;261(1):1–16.
Pellkofer HL, Krumbholz M, Berthele A, Hemmer B, Gerdes LA, Havla J, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology. 2011;76(15):1310–5.
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68(11):1412–20.
Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler. 2011;17(10):1225–30.
Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol. 2016;73(11):1342–8.
Gao F, Chai B, Gu C, Wu R, Dong T, Yao Y, et al. Effectiveness of rituximab in neuromyelitis optica: a meta-analysis. BMC Neurol. 2019;19(1):36.
Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65(11):1443–8.
Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64(7):1270–2.
Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264(9):2003–9.
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19(4):298–306.
Sharman JP, Farber CM, Mahadevan D, Schreeder MT, Brooks HD, Kolibaba KS, et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: results of a phase 2 trial. Br J Haematol. 2017;176(3):412–20.
Konno Y, Kobayashi Y, Takahashi K, Takahashi E, Sakae S, Wakitani M, et al. Fucose content of monoclonal antibodies can be controlled by culture medium osmolality for high antibody-dependent cellular cytotoxicity. Cytotechnology. 2012;64(3):249–65.
Le Garff-Tavernier M, Herbi L, de Romeuf C, Nguyen-Khac F, Davi F, Grelier A, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28(1):230–3.
Zhu W, Zhang Y, Wang Z, Fu Y, Yan Y. Monoclonal antibody-based treatments for neuromyelitis optica spectrum disorders: from bench to bedside. Neurosci Bull. 2020;36(10):1213–24.
Mealy MA, Levy M. A pilot safety study of ublituximab, a monoclonal antibody against CD20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine (Baltimore). 2019;98(25):e15944.
Quách TD, Rodríguez-Zhurbenko N, Hopkins TJ, Guo X, Hernández AM, Li W, et al. Distinctions among circulating antibody-secreting cell populations, including B-1 cells, in human adult peripheral blood. J Immunol. 2016;196(3):1060–9.
Comi G, Bar-Or A, Lassmann H, Uccelli A, Hartung HP, Montalban X, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13–23.
Hammer O. CD19 as an attractive target for antibody-based therapy. MAbs. 2012;4(5):571–7.
Forsthuber TG, Cimbora DM, Ratchford JN, Katz E, Stüve O. B cell-based therapies in CNS autoimmunity: differentiating CD19 and CD20 as therapeutic targets. Ther Adv Neurol Disord. 2018;11:1756286418761697.
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352–63.
Jitprapaikulsan J, Fryer JP, Majed M, Smith CY, Jenkins SM, Cabre P, et al. Clinical utility of AQP4-IgG titers and measures of complement-mediated cell killing in NMOSD. Neurol Neuroimmunol Neuroinflamm. 2020;7(4).
Akaishi T, Takahashi T, Nakashima I, Abe M, Ishii T, Aoki M, et al. Repeated follow-up of AQP4-IgG titer by cell-based assay in neuromyelitis optica spectrum disorders (NMOSD). J Neurol Sci. 2020;410:116671.
Rensel M, Zabeti A, Mealy MA, Cimbora D, She D, Drappa J, et al. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for ⩾4 years in the N-MOmentum trial. Mult Scler. 2021:13524585211047223.
Shi B, Zhao M, Qiao L, Huang F, Zhou S, Wei Y, et al. Relapses shortly after rituximab treatment in neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. 2021;54:103143.
Cree BA, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk D, et al. Sensitivity analysis of the primary endpoint from the N-MOmentum study of inebilizumab in NMOSD. Mult Scler. 2021:1352458521988926.
Marignier R, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk D, et al. Disability outcomes in the N-MOmentum trial of inebilizumab in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2021;8(3).
Patel SY, Carbone J, Jolles S. The expanding field of secondary antibody deficiency: causes, diagnosis, and management. Front Immunol. 2019;10:33.
Pecoraro A, Crescenzi L, Granata F, Genovese A, Spadaro G. Immunoglobulin replacement therapy in primary and secondary antibody deficiency: the correct clinical approach. Int Immunopharmacol. 2017;52:136–42.
Hauser SL, Kappos L, Arnold DL, Bar-Or A, Brochet B, Naismith RT, et al. Five years of ocrelizumab in relapsing multiple sclerosis: OPERA studies open-label extension. Neurology. 2020;95(13):e1854–67.
De La Torre I, Leandro MJ, Valor L, Becerra E, Edwards JC, Cambridge G. Total serum immunoglobulin levels in patients with RA after multiple B-cell depletion cycles based on rituximab: relationship with B-cell kinetics. Rheumatology (Oxford). 2012;51(5):833–40.
Gottenberg JE, Ravaud P, Bardin T, Cacoub P, Cantagrel A, Combe B, et al. Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis Rheum. 2010;62(9):2625–32.
Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol. 2017;14(1):7–18.
Bittner S, Engel S, Lange C, Weber MS, Haghikia A, Luessi F, et al. Diagnostics and treatment of tuberculosis under immunotherapy for multiple sclerosis: current status and recommendations in Germany. Nervenarzt. 2019;90(12):1245–53.
Rheumatology ACo. Medication guide: rituximab [updated February 2020. Available from: https://www.rheumatology.org/Learning-Center/Medication-Guides/Medication-Guide-Rituximab-Rituxan.
Nisar MK, Rafiq A, Östör AJ. Biologic therapy for inflammatory arthritis and latent tuberculosis: real world experience from a high prevalence area in the United Kingdom. Clin Rheumatol. 2015;34(12):2141–5.
Graf J, Leussink VI, Dehmel T, Ringelstein M, Goebels N, Adams O, et al. Infectious risk stratification in multiple sclerosis patients receiving immunotherapy. Ann Clin Transl Neurol. 2017;4(12):909–14.
Ask Mayo Expert, NMOSD medication monitoring guidelines [updated 2/24/2021. Available from: https://askmayoexpert.mayoclinic.org/topic/clinical-answers/gnt-20508358/sec-20508395#jumplinkin3.
Barmettler S, Ong MS, Farmer JR, Choi H, Walter J. Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018;1(7):e184169.
Whittam DH, Tallantyre EC, Jolles S, Huda S, Moots RJ, Kim HJ, et al. Rituximab in neurological disease: principles, evidence and practice. Pract Neurol. 2019;19(1):5–20.
Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82(15):1302–6.
Kieseier BC, Stüve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013;70(3):390–3.
Komai T, Shoda H, Yamaguchi K, Sakurai K, Shibuya M, Kubo K, et al. Neuromyelitis optica spectrum disorder complicated with Sjogren syndrome successfully treated with tocilizumab: a case report. Mod Rheumatol. 2016;26(2):294–6.
Rigal J, Pugnet G, Ciron J, Lépine Z, Biotti D. Off-label use of tocilizumab in neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases: a case-series. Mult Scler Relat Disord. 2020;46:102483.
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72(7):756–63.
Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70(3):394–7.
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020;19(5):391–401.
Gao Y, Zhang B, Yang J. Satralizumab for the treatment of neuromyelitis optica spectrum disorders. Ann Pharmacother. 2021;55(9):1167–71.
Heo YA. Satralizumab: First Approval. Drugs. 2020;80(14):1477–82.
Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114–24.
Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402–12.
Duan T, Tradtrantip L, Phuan PW, Bennett JL, Verkman AS. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology. 2020;162:107827.
Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets. 2017;21(12):1161–70.
Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC, et al. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. Faseb j. 2012;26(5):2197–208.
Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11(6):535–44.
Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol. 2018;9:217.
Juryńczyk M, Jacob A, Fujihara K, Palace J. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol. 2019;19(3):187–95.
Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. 1999;72(1):1–9.
Spadaro M, Winklmeier S, Beltrán E, Macrini C, Höftberger R, Schuh E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. 2018;84(2):315–28.
Stathopoulos P, Chastre A, Waters P, Irani S, Fichtner ML, Benotti ES, et al. Autoantibodies against neurologic antigens in nonneurologic autoimmunity. J Immunol. 2019;202(8):2210–9.
Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. 2017;89(9):900–8.
Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. 2018;90(21):e1858–69.
Lehmann-Horn K, Wang SZ, Sagan SA, Zamvil SS, von Büdingen HC. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight. 2016;1(20):e87234.
Hacohen Y, Kerr W, Waters P. Intrathecal production of MOG-IgG: highlighting the need for CSF testing in clinical practice. Neurology. 2021;97(1):12–3.
Akaishi T, Takahashi T, Misu T, Kaneko K, Takai Y, Nishiyama S, et al. Difference in the source of anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with neuromyelitis optica spectrum disorder. Neurology. 2021;97(1):e1–12.
Macrini C, Gerhards R, Winklmeier S, Bergmann L, Mader S, Spadaro M, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. 2021;144(8):2375–89.
Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139(5):875–92.
Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020;143(5):1431–46.
Fujihara K, Cook LJ. Neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease: current topics. Curr Opin Neurol. 2020;33(3):300–8.
Sechi E, Krecke KN, Messina SA, Buciuc M, Pittock SJ, Chen JJ, et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology. 2021;97(11):e1097–109.
Li X, Wang L, Zhou L, ZhangBao J, Miao MZ, Lu C, et al. The imbalance between regulatory and memory B cells accompanied by an increased number of circulating T-follicular helper cells in MOG-antibody-associated demyelination. Mult Scler Relat Disord. 2019;36:101397.
Winklmeier S, Schlüter M, Spadaro M, Thaler FS, Vural A, Gerhards R, et al. Identification of circulating MOG-specific B cells in patients with MOG antibodies. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):625.
Kaneko K, Sato DK, Nakashima I, Ogawa R, Akaishi T, Takai Y, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry. 2018;89(9):927–36.
Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm. 2019;6(4):e572.
Bruijstens AL, Wendel EM, Lechner C, Bartels F, Finke C, Breu M, et al. E.U. paediatric MOG consortium consensus: part 5 - treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2020;29:41–53.
Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. 2018;75(4):478–87.
Chen JJ, Flanagan EP, Bhatti MT, Jitprapaikulsan J, Dubey D, Lopez Chiriboga ASS, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95(2):e111–20.
Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. 2020;87(2):256–66.
Elsbernd PM, Hoffman WR, Carter JL, Wingerchuk DM. Interleukin-6 inhibition with tocilizumab for relapsing MOG-IgG associated disorder (MOGAD): a case-series and review. Mult Scler Relat Disord. 2021;48:102696.
Zuercher AW, Spirig R, Baz Morelli A, Rowe T, Käsermann F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun Rev. 2019;18(10):102366.
Smith B, Kiessling A, Lledo-Garcia R, Dixon KL, Christodoulou L, Catley MC, et al. Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. MAbs. 2018;10(7):1111–30.
Bril V, Benatar M, Andersen H, Vissing J, Brock M, Greve B, et al. Efficacy and safety of rozanolixizumab in moderate to severe generalized myasthenia gravis: a phase 2 randomized control trial. Neurology. 2021;96(6):e853–65.
Robak T, Kaźmierczak M, Jarque I, Musteata V, Treliński J, Cooper N, et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020;4(17):4136–46.
Acknowledgements
Figures created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Disclosure forms provided by the authors are available with the online version of this article.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Redenbaugh, V., Flanagan, E.P. Monoclonal Antibody Therapies Beyond Complement for NMOSD and MOGAD. Neurotherapeutics 19, 808–822 (2022). https://doi.org/10.1007/s13311-022-01206-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01206-x