
Abstract
At present, there is an increasing interest in the potential role of extracellular vesicles (EVs), acting as multi-signal messengers of the tumor stroma, in the development and progression of tumor. Tumor cell-derived EVs are considered a potential vector for the targeted delivery of antitumor agents due to the ability to fuse with parental cells through endocytosis and release their contents into the cytoplasm of the recipient cell. Tumor cell-derived EVs could be also used for priming immune cells and therapeutic vaccine development. It is also known that mesenchymal stem cells (MSCs) have a tropism toward tumor niches. It is believed that MSC migration to the tumor is due to its inflammatory signaling. Presumably, with the accumulation of MSCs at tumor sites, these cells differentiate into pericytes or tumor-associated fibroblasts, thereby forming a supporting tumor growth microenvironment. However, besides the ability to promote tumor progression, MSCs can also suppress its growth by inhibiting proliferation and cell cycle progression, and angiogenesis. Thus, the further studies of the MSC role in TME and MSC interaction with other cells of the tumor stroma, including through EVs, are of particular interest. To increase the yield of vesicles the isolation method based on pharmacological disorganization of the actin cytoskeleton induced by treating with cytochalasin B was used in this study. In this investigation the interaction of SH-SY5Y neuroblastoma cell-derived membrane vesicles, obtained using cytochalasin B (CIMVs), with human bone marrow-derived MSCs was analyzed using imaging flow cytometry. Using transmission electron microscopy, it was shown that CIMVs have a size similar to that of natural microvesicles, which is 100–1000 nm. Using imaging flow cytometry, it was shown that after 24 h of co-cultivation 6% of the MSCs contained a large number of CIMVs, and 42% of the MSCs contained a small amount of CIMVs. Cultivation of MSCs with SH-SY5Y cell-derived CIMVs also induced dose-dependent decrease in the expression of CD markers typical for MSCs. Thus, the internalization of SH-SY5Y cell-derived CIMVs within MSCs and the ability of the CIMVs to modulate immunophenotype of the recipient cells were shown. However, further studies are required to determine the effect of CIMVs on pro- or antioncogenic phenotype and function of MSCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Due to the key role in intercellular communication, considerable interest is directed to the study of extracellular vesicles (EVs), membrane vesicles secreted into the extracellular environment by almost all cell types of the body. Depending on the cellular origin, EVs can be classified into exosomes, microvesicles (MVs) and apoptotic bodies (ABs) [1]. Exosomes are formed by endocytosis. MVs are released by direct budding and fusion of the plasma membrane [2]. Both exosomes and MVs carry the cargo of the parental cell represented by various proteins and nucleic acids. Unlike other types of EVs, ABs are formed only when cells are fragmented during apoptosis [1].
At present, there is an increasing interest in exploring the potential role of EVs in tumor development and progression. Besides tumor cells, the tumor microenvironment (TME) contains a number of other cell types, such as fibroblasts, epithelial and endothelial cells, lymphocytes, adipocytes, inflammatory cells, and mesenchymal stem/stromal cells (MSCs) [3]. EVs act as key mediators in maintaining and spreading the tumor by mediating intercellular communication in TME and thus inducing phenotypic modifications of the recipient cells [4]. Tumor cells, among other ways, use EV-mediated intercellular signaling to maintain tumor vascularization, invasion, and immune escape, as well as to form an aggressive phenotype and drug resistance [1]. EVs can carry oncogenes and onco-miRNAs, thereby modulating the activity of various signaling pathways in recipient cells and playing a crucial role in tumor growth and metastasis [5].
MSCs if released to TME can be differentiated into myofibroblasts [6], tumor-associated fibroblasts (TAMs) [7], or pericytes, thus supporting tumor growth [8], while also MSCs can induce epithelial-mesenchymal transition (EMT), a process in which epithelial cells lose their polarity and adhesiveness and acquire migratory and invasive properties [14, 18, 19].
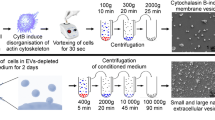
Thus, the further studies of the MSC role in TME and MSC interaction with other cells of the tumor stroma, including through EVs, are of particular interest. In order to increase the yield of obtained vesicles in this work, the isolation method based on pharmacological disorganization of the actin cytoskeleton by treating the cells with cytochalasin B and vortexing was used. Cytochalasin B allows obtaining nucleus-free vesicles, surrounded by a cytoplasmic membrane and containing the content of parental cell cytoplasm. A distinctive feature of cytochalasin B-induced membrane vesicles (CIMVs) is the lack of active mechanism of cargo sorting during CIMV generation [20].
This paper examines the interaction of CIMVs isolated from tumor neuroblastoma cells with bone marrow-derived MSCs. For the first time, CIMVs obtained from neuroblastoma SH-SY5Y cells were characterized using transmission electron microscopy and flow cytometry, as well as the interaction of CIMVs from tumor neuroblastoma cells with MSCs was assessed using imaging flow cytometry. Also, the influence of tumor cell-derived CIMVs on the immunophenotype of bone marrow-derived MSCs was investigated.
2 Materials and Methods
2.1 Cells and Culture Conditions
Bone marrow samples were provided by the Republican Clinical Hospital for research purposes in accordance with ethical standards and current legislation (the protocol was approved by the Committee on Biomedical Ethics of Kazan Federal University (No. 3, 03/23/2017)). Written informed consent was obtained from donors. MSCs from human bone marrow were isolated in a ficoll density gradient (1.077 g/cm3, PanEco, Russia), followed by adhesion of cells to the surface of culture plastic as previously described [21]. SH-SY5Y human neuroblastoma cells were obtained from the American Type Culture Collection (ATCC number: CRL-2266, Manassas, VA, USA). MSCs and SH-SY5Y were cultured in DMEM/F12 (PanEco, Russia) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, USA), 2 mM L-glutamine (PanEco, Russia), penicillin (100 U/ml), and streptomycin (100 µg/ml) (Biolot, Russia). Cells were maintained at 37 °C in a humid atmosphere with a content of 5% CO2. Sub-confluent cells (approximately 80% confluent) were passaged using 0.25% trypsin–EDTA solution (Invitrogen, USA).
2.2 Immunophenoty**
MSC immunophenoty** was performed using the antibodies to CD29 (#87,106, Sony, USA), CD44 (#51–9,007,656, BD Biosciences, USA), CD73 (#51–9,007,649, BD Biosciences, USA), CD90 (#51–9,007,657, BD Biosciences, USA), and CD105 (#323,218, BioLegend, USA); a cocktail of PE-conjugated antibodies hMSC Negative Cocktail from the Human MSC Analysis Kit (#562,245, BD Biosciences, USA) was used as a negative control to identify potential contaminants with hematopoietic cells [22, 23]. The staining results were analyzed using FACS Aria III (BD Biosciences, USA) and BD FACSDiva™ software version 7.0.
2.3 CIMV Production
CIMVs were isolated as previously described [24]. SH-SY5Y cells were washed twice with Dulbecco’s Phosphate-Buffered Saline (DPBS, PanEco, Russia) and incubated in DMEM/F12 without serum containing 10 μg/ml of cytochalasin B (Sigma-Aldrich, USA) for 30 min (37 °C, 5% CO2). At the end of incubation, the cell suspension was vortexed vigorously for 30 s and centrifugated (500 g for 10 min). The supernatant was subjected to two subsequent centrifugation steps (700 g for 10 min and 12000 g for 15 min). The resulting pellet contained CIMVs from SH-SY5Y cells. Total protein concentration in CIMV samples was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, USA) according to manufacturer’s recommendations. The results were measured at wavelength 562 nm.
To determine the size of the isolated CIMVs, flow cytometry analysis was performed using SPHERO™ Nano Fluorescent Particle Size Standard Kit (Spherotech, USA).
2.4 Analysis of CD Marker Expression in the CIMVs
CIMVs were isolated from 1 × 106 SH-SY5Y cells, washed once with PBS, and stained with 1 µl of anti-CD63 (PerCP/Cy5.5) (#353,020, BioLegend, USA), anti-CD81 (PE/Cy7) (#349,512, BioLegend, USA), and anti-tumor susceptibility 101 (TSG101) (PE) (#ab209927, Abcam, UK) antibodies against specific surface markers for 30 min in the dark at room temperature (RT). After that, CIMVs were washed twice with PBS and analyzed using flow cytometer FACS Aria III (BD Biosciences, USA) and BD FACSDiva™ software version 7.0. To analyze the presence of intracellular markers heat shock protein 70 kDa (Hsp70) and calnexin, newly isolated CIMVs were fixed with 0.01% formaldehyde for 15 min at RT and washed with PBS for 5 min. Next CIMV membranes were permeabilized using 0.1% Triton X-100 for 15 min at RT. After that, the samples were washed twice with PBS and stained with anti-Hsp70 (FITC) (#648,004, BioLegend, USA) and anti-Calnexin (Alexa Fluor 594) (#ab203439, Abcam, UK) antibodies for 30 min in the dark at RT. Stained vesicles were washed twice with PBS and analyzed using FACS Aria III (BD Biosciences, USA) and BD FACSDiva™ software version 7.0.
2.5 Transmission Electron Microscopy
CIMVs were fixed in 2.5% glutaraldehyde on PBS solution (12 h at 4 °C), and then washed twice with phosphate buffer by centrifugation, and precipitated vesicles were resuspended in 100 μl of phosphate buffer. The resulting solution (5 μl) was dropped on nickel grids coated with Formvar-Carbon film (200 mesh, Sigma-Aldrich, USA), and the sample were contrasted with uranyl acetate and lead citrate. The grids were dried for 12 h in air at 37 °C. Samples were examined using transmission electron microscope HT7700 (Hitachi, Japan) (accelerating voltage: 100 kV) at the KFU Interdisciplinary Center for Analytical Microscopy.
2.6 Cytoplasmic Membrane Staining
Fluorescent cell membrane staining of MSCs and CIMVs was performed in a serum-free medium with Vybrant Cell Labeling Solutions kit (Thermo Fisher Scientific, USA) [21, 25] using DiO (green emission spectrum) and DiD (red emission spectrum) dyes, respectively, according to manufacturer’s recommendations. A qualitative assessment of staining was performed on an Axio Observer.Z1 fluorescence microscope (Carl Zeiss, Germany) using AxioVision Rel. 4.8 software.
2.7 Imaging Flow Cytometry
After fluorescent staining, 1 × 106 MSCs were seeded onto 6-well plate and cultured in full DMEM/F12 medium containing 100 μg/ml of CIMVs for 24 h at 37 °C in 5% CO2. After that, cells were trypsinized and washed with DPBS three times by centrifugation (at 500 g for 5 min). Cell pellets were resuspended in DPBS. Imaging flow cytometry was performed using the 5-laser ImageStreamX Mark II (Amnis-EMD-Millipore, USA). Data were collected using Inspire software and further analyzed by Ideas software.
3 Results and Discussion
Currently, there is an increasing interest in the role of MSCs in tumor progression. One of the key factors in this process is the modulation of MSC properties through tumor cell-derived EVs. In this study, MSCs were isolated from human bone marrow. Immunophenotypic analysis showed that isolated MSCs had a mesenchymal stem cell phenotype and expressed surface markers CD29 (99.9%), CD44 (100%), CD73 (79.8%), CD90 (99.5%), and CD105 (91.3%), while the expression of hematopoietic cell surface markers (CD11b, CD19, CD34, CD45, HLA-DR) was insignificant (Fig. 1).

Immunophenotypic characteristics of human bone marrow MSCs (passage 0). Negative markers, hematopoietic cell markers: CD34, CD11b, CD19, CD45, and HLA-DR
There are several methods to isolate natural EVs from cell cultures. One of the most common methods is differential centrifugation, in which particles are separated based on their different densities or sizes. For additional purification, an extra step using sucrose or iodixanol cushion/gradient can be applied. Another common method for EV isolation is ultrafiltration, in which EVs are separated based on their size [26]. In addition to the abovementioned methods for the isolation of EVs polymer-based precipitation, size-exclusion chromatography and immunoaffinity are also used. Every approach of EV isolation has its advantages and disadvantages [Full size image
