
Abstract
In Parkinson’s disease, the dysfunction of the dopaminergic nigrostriatal tract involves the loss of function of dopaminergic neurons of the substantia nigra pars compacta followed by death of these neurons. The functional recovery of these neurons requires a deep knowledge of the molecules that maintain the dopaminergic phenotype during adulthood and the mechanisms that subvert their activity. Previous studies have shown that transcription factor NURR1, involved in differentiation and maintenance of the dopaminergic phenotype, is downregulated by α-synuclein (α-SYN). In this study, we provide a mechanistic explanation to this finding by connecting α-SYN-induced activation of glycogen synthase kinase-3 (GSK-3) with NURR1 phosphorylation followed by proteasomal degradation. The use of sequential deletion mutants and single point mutants of NURR1 allowed the identification of a domain comprising amino acids 123-PSSPPTPSTPS-134 that is targeted by GSK-3 and leads to subsequent ubiquitination and proteasome degradation. This study provides a detailed analysis of the regulation of NURR1 stability by phosphorylation in synucleinopathies such as Parkinson’s disease.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Midbrain dopaminergic (DAergic) neurons are the main source of dopamine (DA) in the mammalian central nervous system. Several transcription factors have been implicated in DAergic differentiation [1]. Among them, nuclear receptor‐related factor 1 (NURR1; also known as NR4A2) is a transcription factor of the orphan nuclear receptor class that participates in acquisition of the DAergic phenotype in neurons during development and in the maintenance of their functionality during adulthood [2,3,4]. It regulates the expression of several genes involved in DA metabolism, including tyrosine hydroxylase (TH) [5,6,7], dopamine transporter (DAT) [8], amino acid decarboxylase (AADC) [9], vesicular monoamine transporter-2 (VMAT2) [9], as well as other non-DAergic genes such as NRP1 [10] and RET (GDNF receptor) [11]. Ablation of NURR1 in adult rodents results in reduced expression of its target genes and loss of functional DAergic midbrain neurons [12,13,14]. Mutations in the human NURR1 gene have been identified in association with Parkinson’s disease (PD), where neurodegeneration of the DAergic neurons of the SN occurs [15, 16].
DAergic neuronal loss is associated with abnormal accumulation and aggregation of the protein α-synuclein (α-SYN) in the form of Lewy bodies and Lewy neurites [17]. Point mutations, duplications, and triplications of α-SYN gene (SNCA) are associated with familiar forms of PD [18, 19], which indicate a key role of this protein in the neurodegenerative process of the disease. The toxicity elicited by α-SYN oligomers correlates with its phosphorylation at serine 129 as this event promotes fibril formation [20, 21]. Related to NURR1, a seminal study, demonstrated that aberrant expression of human α-SYN in murine DAergic neurons correlates with exacerbated proteasomal degradation of NURR1 and loss of the DAergic phenotype [22]. However, little is known about the molecular mechanisms that connect synucleinopathy with loss of NURR1 stability and loss of DAergic neuron functionality, which may be more important than frank cell loss [3, 12]. This information is crucial to identify early signs of damaged DAergic neurons and to apply a neuroprotective therapy before the manifestation of DAergic cell death.
Phosphorylation is a mechanism used in many proteins to target their proteolytic degradation. For instance, glycogen synthase kinase 3 (GSK-3) phosphorylates several proteins to create a phosphorylation-dependent degradation domain (phosphodegron) that is then recognized by a variety of E3 ubiquitin ligase adapters leading to proteasomal degradation of the phosphorylated protein [23]. Considering that GSK-3 is activated by α-SYN aggregates [24,25,26,27,28], here, we analyzed if NURR1 is phosphorylated by GSK-3 and sent to ubiquitin proteasome degradation phosphorylation by GSK-3. The two isoforms of GSK-3 (GSK-3α and GSK-3β) play critical roles in metabolism, neurogenesis, proliferation, neuronal differentiation, and neuronal death [29], and their dysregulation is associated with neurodegenerative diseases. For instance, abnormal GSK-3β activity leading to TAU phosphorylation and aggregation has been extensively reported in Alzheimer’s disease [30,31,32] but also in connection with several hallmarks of PD [33,34,35,36]. Thus, in postmortem PD brain samples, GSK-3β activity is increased in regions related to PD pathology, and GSK-3β co-localizes with α-SYN in Lewy bodies [35, 36]. α-SYN pathology leads to GSK-3β activation, subsequent phosphorylation of several transcription factors such as Jun, Myc, HSF-1, and CREB, and neuronal death, thus opening the possibility of a similar regulation of NURR1 [28].
In this study, using several models of synucleinopathy, we found that α-SYN-induced activation of GSK-3β leads to phosphorylation of NURR1 and its subsequent ubiquitin–proteasome degradation, which precedes loss of the DAergic phenotype.
Materials and Methods
A detailed description of methods is presented in Supplemental Material
Cell Culture and Reagents
Human embryonic kidney 293 T with SV40 T antigen (HEK293T) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, Madrid, Spain) supplemented with 10% fetal bovine serum (Invitrogen, CA, USA) and 80 µg/ml gentamycin (Gibco, MA, USA). Human neuroblastoma cells (SH-SY5Y) were cultured in RPMI supplemented with 10% fetal bovine serum (Invitrogen) and 80 µg/ml gentamicin. The SH-SY5Y-α-SYN Tet-Off cells, described previously [37, 38], were cultured in RPMI with 10% fetal bovine serum, 250 µg/ml G418 (Gibco), 50 µg/ml hygromycin B (Invitrogen), and 2 µg/ml doxycycline (DOX) (Sigma-Aldrich). The expression of α-SYN was switched on by DOX removal. Transient transfection of HEK293T cells was performed with TransFectin Lipid Reagent (Bio-RAD, CA, USA). The inhibitors SB216763, LY294002, and MG132 were from Sigma-Aldrich. Cycloheximide (CHX) was from Boehringer Mannheim (Stuttgart, Germany).
Plasmids and Lentiviruses
The vectors pCGN-HA-GSK-3βΔ9, pCGN-HA-GSK-3βWT, and pCGN-HA-GSK-3βY216F were provided by Dr. Akira Kikuchi (Department of Biochemistry, Faculty of Medicine, Hiroshima University). Vectors pGL3-NBRE3xLuc and pGL3-TkLuc were provided by Dr. Philippe Lefebvre (INSERM Institut Pasteur de Lille, Lille, France). The HA-Ubiquitin expression vector was provided by Dr. Tadashi Nakagawa (Division of Cell Proliferation, ART, Tohoku University Graduate School of Medicine, Sendai, Japan). The plasmid pcDNA3.1-Nurr1WT-V5/6xHis has been described previously [39]. The lentiviral particles used in this study were purchased from Addgene and were generated in HEK293T cells as described previously ([40] and Supplemental Material).
α-SYN Pre-formed Fibrils (PFF)
Purified monomeric α‐SYN was purchased from Proteos, Inc (cat no. RP‐003), and PFFs were formed according to the protocol provided by the manufacturer [41, 42] (see Supplemental Material).
Immunoblotting
This protocol was performed as described in [43]. Briefly, cells were homogenized in lysis buffer (TRIS pH 7.6 50 mM, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% SDS), and samples were heated at 95 °C for 15 min, sonicated and pre-cleared by centrifugation. Proteins were resolved in SDS-PAGE and transferred to Immobilon-P (Merck-Millipore, MA, USA) membranes. Proteins of interest were detected with the primary antibodies indicated in the Supplemental Table 2. Proper peroxidase-conjugated secondary antibodies were used for detection by enhanced chemiluminescence (GE Healthcare).
Immunofluorescence
SH-SY5Y cells were seeded in 24-well plates (5 × 103 cells/well) on poly-D-Lys-covered slides and treated with 1 µg/ml PFFs. The protocols have been previously described [39, 44, 45]. Primary antibodies recognized TH (Merck-Millipore), human α-SYN (Santa Cruz Biotechnology, Dallas, TX, USA) and α-SYN-pSer129 (Abcam, Cambridge, UK). Secondary antibodies were as follows: Alexa Fluor 488 donkey anti-mouse, and Alexa 546 donkey anti-rabbit (1:500; Thermo Fisher Scientific, Waltham, MA, USA) and Alexa Fluor 546-conjugated donkey anti-mouse IgG (Molecular Probes, Eugene, OR, USA). Control sections were treated identically but omitting the primary antibody.
In vivo Ubiquitination Assay
HEK293T cells co-transfected with expression plasmids for HA-Ubiquitin (HA-Ub), Nurr1WT-V5/6xHis, or Nurr1MUT2-V5/6xHis with pCGN-HA-GSK-3βΔ9 or pCGN-HA-GSK-3βY216F, using TransFectin Lipid Reagent (Bio-RAD). After 5 h, HEK293T cells were treated for 16 h with 2 μM MG132 (Sigma-Aldrich). Cells were then lysed in a RIPA buffer (150 mM NaCl, 25 mM Tris–HCl, pH 7.5, 1% Nonidet P-40, 1% sodium deoxycholate, 1% Triton-X100, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 g/ml leupeptin). Then, samples were kept for 30 min at 4 °C in a rotating wheel and centrifuged at 13,000 rpm for 10 min. Three microliters of the anti-V5 (Invitrogen) was added per lysate, and after incubation for 2 h at 4 °C in a rotating wheel, gamma-bind Sepharose-protein G was added (Amersham Biosciences), followed by incubation for 1 h at 4 °C. The complexes were harvested by centrifugation and washed three washes with RIPA buffer, resolved in SDS–polyacrylamide gels, and immunobloted. Mouse IgG TrueBlot (eBiosciences) was used as a peroxidase-conjugated secondary antibody (1:10,000 dilution) because it reduces interference by the 55-kDa heavy and 23-kDa light chains of the immunoprecipitation antibody.
Lambda Phosphatase Assay
HEK293T cells were co-transfected with Nurr1WT-V5/6xHis and pCGN-HA-GSK-3βΔ9 or pCGN-HA-GSK-3βY216F using TransFectin Lipid Reagent (Bio-Rad) according to manufacturer recommendations. After 24 h of recovery from transfection, the cells were lysed in 200 μl lysis buffer (137 mM NaCl, 20 mM Tris–HCl, pH 7.5, 1% Nonidet P40, 10% glycerol, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). Then, the samples were sonicated and precleared by centrifugation, and 50 μl of the sample was incubated with λ-protein phosphatase (Upstate, Millipore) for 4 h at 37 °C. Then, the samples were resolved by SDS-PAGE and immunoblotted.
Two-Dimensional Electrophoresis
HEK293T cell co-transfected with expression plasmids for Nurr1WT-V5/6xHis, Nurr1MUT2-V5/6xHis and pCGN-HA-GSK-3βΔ9 or pCGN-HA-GSK-3βY216F, using TransFectin Lipid Reagent (Bio-RAD) according to manufacturer recommendations. For experimental details, see Supplemental Material.
Analysis of mRNA Levels
Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). Reverse transcription and quantitative PCR were done as detailed elsewhere [44]. Primer sequences are shown in Supplemental Table 3. Data analysis was based on the ΔΔCT method with normalization of the raw data to housekee** genes Actb and Gapdh (Applied Biosystems, Thermo Fisher Scientific). All PCRs were performed in triplicate.
Luciferase Assays
Luciferase activities were determined using a luciferase assay system (Promega) as per the manufacturer’s instructions. As a reference plasmid to normalize transfection efficiency, a CMV-galactosidase plasmid (Promega) was cotransfected in all experiments and luciferase assay values were normalized to galactosidase activity.
Statistics
Results are expressed as mean ± SEM from at least three independent experiments. Data were analyzed by one-way ANOVA followed by Newman–Keuls multiple comparison test (p ≤ 0.001), or with Student’s t test (p ≤ 0.05), using Prism version 5.03 software (GraphPad, San Diego, CA, USA).
Results
α-SYN Aggregates Reduce the DAergic Phenotype of SH-SY5Y Cells
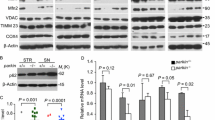
In order to identify the mechanism involved in the dysregulation of the DAergic phenotype, we incubated the DAergic cell line SH-SY5Y with preformed fibrils (PFFs) of human recombinant α-SYN (1 µg/ml, 10 days). Confocal microscopy demonstrated the formation of aggregates containing α-SYN and Ser129-phopshorylated α-SYN (α-SYN-pSer129) (Fig. 1A). PFFs induced a slight nonsignificant decrease in NURR1 transcript levels compared to the control untreated cells, indicating similar NURR1 gene expression, but at the same time, TH and RET transcripts were diminished (Fig. 1B). By contrast, not only TH and RET proteins were decreased but also NURR1 (Fig. 1C and D). The fact that NURR1 gene expression was little or no affected by PFFs (see “Discussion”), together with the decrease in NURR1 protein levels, suggests that α-SYN PFFs must cause, at least in part, a reduction of NURR1 protein stability and subsequent decrease in the expression of NURR1 target genes, such as TH and RET.

Pre-formed fibrils (PFF) of α-SYN disturb the DAergic phenotype. A Confocal immunofluorescence of SH-SY5Y cells submitted to PFF (1 µg/ml PFF, 10 days). Note the presence of cytoplasmic α-SYN-pSer129 aggregates in the presence of PFFs. B qRT-PCR determination of transcript levels for NURR1, TH, and RET normalized by the average of housekee** genes ACTB and GAPDH. Values are mean ± S.E.M. (n = 3). Statistical analysis was performed with Student’s t test. *p < 0.05 vs. untreated cells. C Immunoblots of cells treated under the same conditions. Upper two panels, anti-α-SYN-p-Ser129 and anti-total α-SYN antibodies. Lower four blots, anti-NURR1, anti-TH, anti-RET, and-GADPH antibody used as a protein loading control. D Densitometric quantification of NURR1, TH, and RET protein levels relative to GAPDH is representative blots of C. Data are mean ± SEM (n = 3). Statistical analysis was performed with Student’s t test. *p < 0.05 vs. untreated cells
We further analyzed the regulation of NURR1 in the Tet-Off SH-SY5Y cell line, conditionally expressing α-SYN in the absence of doxycycline (DOX) [Full size image
GSK-3 Is Needed to Downregulate NURR1
α-SYN overexpression reduced the levels of GSK-3β phosphorylated at Ser9 (GSK-3β-pSer9) (Fig. 2A, B). This phosphoserine exerts an inhibitory effect on the kinase by blocking the catalytic site [47, 48]. Therefore, reduction in pSer9-GSK-3 levels is indicative of increased kinase activity. To determine if increased GSK-3 activity might affect NURR1 stability, we first used the easy-to-transfect human cell line HEK293T. Transcriptomic data indicate that these cells originated from the neural crest and express several neuron-specific genes [49, 50]. We ectopically expressed a V5-targed NURR1 together with constitutively active GSK-3β lacking the first nine amino-terminal residues that correspond to the pseudosubstate (GSK-3βΔ9). Lack of Ser9 renders this kinase insensitive to downregulation by AKT-mediated phosphorylation. As a negative control, we used a hypomorphic mutant containing a single point Tyr-to-Phe mutation in its activation loop (GSK-3βY216F) that renders this kinase almost inactive [51, 52]. As shown in (Fig. 3A), NURR1 levels were reduced with increasing amounts of the GSK-3βΔ9, and were not altered in the presence of inactive GSK-3βY216F. Then, we silenced the expression of both α and β isoforms of this kinase by lentiviral knock-down for 3 days in V5-NURR1 expressing HEK293T cells (Fig. 3B) as well as in naïve SH-SY5Y cells (Fig. 3C). As a control, we analyzed β-catenin levels, a well-established substrate of GSK-3, which is degraded upon GSK-3β-mediated phosphorylation. Just a ~ 50% decrease in GSK-3α and GSK-3β levels increased NURR1 levels and its targets RET and TH. We also silenced both isoforms in the Tet-Off SH-SY5Y-α-SYN cells (Fig. 3D-G). Despite α-SYN overexpression, GSK-3α and GSK-3β knock-down rescued NURR1 levels to baseline as well as TH and RET levels. The effect was most evident in the GSK-3β knocked-down cells, indicating a preponderant role for this isoform. These results show for the first time that GSK-3 is required for the downregulation of NURR1 induced by α-SYN and are in line with observations based on GSK-3 inhibitors in the Parkinsonian MPTP and MPP+ [53, 54], or 6-OHDA [55,56,57] models. The mechanisms of action of these two toxins, altering mitochondrial activity and redox homeostasis, is at least partially different to the proteinopathy elicited by α-SYN aggregates, therefore suggesting that these different pathomechanisms overlap on the GSK-3/NURR1 axis reported here for α-SYN.

GSK-3 is instrumental in α-SYN-induced NURR1 downregulation. A Reduction of NURR1 levels in the presence of of GSK-3β. HEK293T cells were co-transfected with the NURR1WT-V5 expression vector and the indicated amounts of the HA-GSK-3βY216F mutant and dominant-positive HA-GSK-3βΔ9 mutant, and then maintained in low-serum medium for 16 h. GFP expression was used as control for even transfection. Whole-cell lysates were immunoblotted against anti-V5 antibody (NURR1) or anti-HA antibody (GSK-3β). β-Catenin levels were immunoblotted as a control for GSK-3 activity and anti-GAPDH antibody as control of protein load. B HEK293T cells were infected with an expression vector for V5-tagged NURR1 and the lentiviruses specific shCtrl, shGSK-3α, and shGSK-3β. Upper blots, V5-NURR1 and β-catenin protein levels. Lower blots, GSK-3α/β and GAPDH protein levels. C Naïve SH-SY5Y cells were infected with lentiviruses that carried a specific silencer for a control scramble sequence (shCtrl), GSK-3α (shGSK-3α) or GSK-3β (shGSK-3β). After 24 h, cells were transferred to Opti-MEM Reduced Serum Medium, and grown for 3 days. Upper blot, NURR1 protein levels. β-Catenin levels were used as positive control of GSK-3 activity. In the middle blots, GSK-3α/β protein levels as control shRNA treatment, and TH and RET protein levels as NURR1 targets. Lower blot, GAPDH protein levels showing similar protein load per lane. D Tet-Off SH-SY5Y cells were treated with vehicle or DOX (2 µg/ml, 5 days) in RPMI with serum. Then, the cells were infected with shCtrl, shGSK-3α and shGSK-3β lentiviruses. After 24 h, cells were transferred to Opti-MEM Reduced Serum Medium for 8 days maintaining the DOX treatment. Upper panels, tetracycline-induced α-SYN and its α-SYNpSer129. Middle panels, expression of endogenous NURR1, TH and RET and GAPDH. Lower panels, levels of GSK-3β-pSer9 and total levels of GSK-3α/β. E, F, G Quantitative determination of NURR1, RET, and TH levels, respectively, normalized by GADPH levels. Values represent mean ± SEM (n = 3). A Student’s t test was used to assess difference among groups. *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant
GSK-3 Targets NURR1 for Degradation
We next examined the effect of GSK-3 on NURR1 turn-over. SH-SY5Y cells were infected with lentiviral vectors expressing shCTRL, shGSK-3α/shGSK-3β. After 3 days, cells were incubated with the protein synthesis inhibitor cycloheximide (CHX, 100 µM) (Fig. 4A, B). As a control, we monitored the stability of β-catenin. In shCTRL cells, NURR1 had a half-life of ~ 6 h. However, in the GSK-3-knocked-down cells NURR1 was almost completely stable during this time. As an additional approach, we performed similar experiments but inhibiting GSK-3 with the potent and selective inhibitor SB216763 (5 µM, pre-incubated for 2 h) (Fig. 4C, D). In vehicle-treated cells, NURR1 exhibited a half-life of ~ 6 h, as observed before, but in SB216763-treated cells, the levels of NURR1 were hardly affected. Therefore, both genetic and chemical inhibition of GSK-3 results in stabilization of NURR1.

GSK-3 reduces the half-life of NURR1. A SH-SY5Y were infected with lentiviral silencers shCtrl, shGSK-3α, and shGSK-3β in RPMI with serum. After 24 h, cells were transferred to Opti-MEM Reduced Serum Medium for 3 days. Finally, the cells were treated with 100 µM CHX for the indicated times. Upper blots, NURR1 protein levels, and β-catenin protein levels used as positive control GSK-3 activity. Middle blots, GSK-3α/β protein levels, as control shRNA knock-down. GAPDH protein levels, as protein loading control. B Graph depicts the natural logarithm of the relative levels of the NURR1 protein as a function of CHX chase time in SH-SY5Y cells treated like in A. C Cells were maintained in Opti-MEM Reduced Serum Medium for 16 h and then treated with the GSK-3 inhibitor SB216763 (5 μM, 2 h) prior to inhibition of protein synthesis with CHX. D Graph depicts the natural logarithm of the relative levels of the NURR1 protein as a function of CHX chase time in SH-SY5Y cells treated like in C. For B and D, the protein half-life was determined in the linear range of the degradation curve. Statistical analysis was performed with one-way ANOVA followed by Newman–Keuls multiple comparison test. ***p < 0.001
GSK-3β Targets NURR1 for Phosphorylation and Degradation Through the UPS
HEK293T cells were co-transfected with expression vectors for NURR1-V5 and HA-GSK-3βΔ9, or HA-GSK-3βY216F as control. After 16 h, the cells were treated for 2 h and 4 h with the selective proteasome inhibitor MG132 (20 µM). As shown in Fig. 5A, UPS inhibition protected NURR1 from GSK-3β-mediated degradation. In an ubiquitination assay, HEK293T cells were co-transfected with expression vectors for NURR1-V5 along with HA-tagged ubiquitin and either HA-GSK-3βY216F, or HA-GSK-3βWT or HA-GSK-3βΔ9 (Fig. 5B, C). Overexpression of GSK-3βWT slightly ubiquitylated NURR1, and constitutively active GSK-3βΔ9 considerably enhanced ubiquitination. It has been reported previously that NURR1 is degraded by the UPS [22, 58, 59], but our results show for the first time the direct participation of GSK-3β.

GSK-3 induces NURR1 phosphorylation and degradation. A HEK293T cells were co-transfected with expression vectors for NURR1-V5 and hypomorphic HA-GSK-3βY216F or active HA-GSK-3βΔ9. After 16 h, cells were subjected to the ubiquitin–proteasome inhibitor MG132 (20 µM, for 2 h and 4 h). Upper blot, anti-V5 antibody showing ectopically expressed NURR1-V5; middle panel, anti-HA antibody showing the GSK-3β proteins. Note the smaller size of HA-GSK-3βΔ9 due to the deletion of the first nine N-terminal residues. Lower blot, GAPDH levels showing similar protein load per lane. B, C Ubiquitination assay. HEK293T cells were co-transfected with the indicated plasmids and HA-tagged ubiquitin (HA-Ub) expression vector. One fifth of whole-protein lysate was used as input to control for protein expression (B). The rest of the protein lysates were immunoprecipitated with anti-V5 antibody and immunoblotted with anti-HA (Ub) indicated in C. D Schematic illustration of NURR1-V5 chimeras used for map** GSK-3-sensitive sites in NURR1. EGFP, enhance green fluorescent protein; A/B (AF1), DBD, LBD (AF2), V5, C-terminal tag used for detection in immunoblot. E HEK293T cells were co-transfected with EGFP-NURR1WT-V5 and the chimeric deletion mutants, along with HA-GSK-3βY216F or HA-GSK-3βΔ9 expression vectors, and then maintained in low-serum medium for 16 h. Whole-cell lysates were immunoblotted against anti-V5 antibody (EGFP-NURR1 chimeras) or anti-HA antibody (GSK-3β). Lamin B levels show similar protein load per lane. F Amino acid sequence of Core2 in wild-type NURR1 and the alanine substitutions mutated to generate NURR1MUT2-V5. G HEK293T cells were co-transfected with NURR1WT-V5 and NURR1MUT2-V5, along with HA-GSK-3βY216F or HA-GSK-3βΔ9 as indicated. Then, cells were maintained in low-serum medium for 16 h before immunoblotting with the indicated antibodies
GSK-3 phosphorylates its substrates in two specific consensus sequences, (Ser/Thr)-Pro or (Ser/Thr)-X3-(pSer/pThr), where X is any residue [60]. Using the NetPhos 2.0 program, we found that NURR1 contains at least five putative sequences with serines or threonines that conform to the consensus motif for GSK-3 phosphorylation. We named these sites, Core 1, 2, 3, 4, and 5 (Fig. 5D and Supplemental Fig. 1A). Then, we generated sequential deletion mutants fused to enhanced green fluorescence protein (EGFP) at the N-terminus and a V5 tag at the C-terminus (Fig. 5D) and analyzed their phosphorylation pattern (Fig. 5E). GSK-3βΔ9 induced a slightly retarded EGFP-NURR1 band in the full length chimera and in the first deletion mutant (Δ1), compared to control GSK-3βY216F. In addition, the protein levels of these two constructs were decreased in the presence of GSK-3βΔ9. By contrast, the rest of the EGFP-NURR1 mutants exhibited low or no obvious band shift and were resistant to degradation in the presence of GSK-3βΔ9, suggesting that the sites targeted by GSK-3β on NURR1 are preferentially located before or at Core 2 (Fig. 5E). As control, EGFP alone was insensitive to GSK-3β-induced band shift or degradation (data not shown). Additionally, we performed point mutations of NURR1 at Core 2 by changing 4 serines and 2 threonines to alanines (Fig. 5F). We found that Core 2 mutation rendered NURR1 insensitive to GSK-3β-induced band shift and degradation (Fig. 5G). The amino acid sequence of Core 2, comprising residues 123 to 134 is highly conserved in vertebrates (Supplemental Fig. 1B-C).
To confirm that GSK-3β induces NURR1 phosphorylation, we performed a lambda-phosphatase assay (λPPase) in HEK293T cells co-transfected with expression vectors for NURR1-V5 and GSK-3βΔ9 or GSK-3βY216F (Fig. 6A). In the presence of GSK-3βΔ9, NURR1 showed a retarded band in SDS-PAGE. This gel shift was abrogated when the protein lysate was incubated with the phosphatase, therefore demonstrating that the retarded band is due to GSK-3-mediated phosphorylation.

GSK-3 induces the phosphorylation of NURR1. A Lambda phosphatase assay. Cells were co-transfected with expression vector for NURR1-V5 and either HA-GSK-3βY216F or HA-GSK-3βΔ9. Cell lysates were incubated with or without λ-phosphatase as indicated. Empty arrowhead, retarded band that corresponds to phosphorylated NURR1; black arrowhead, non-phosphorylated NURR1. B Analysis of NURR1 phosphorylation by 2D-PAGE. HEK293T cells were co-transfected with NURR1WT-V5 and NURR1MUT2-V5, along with HA-GSK-3βY216F or HA-GSK-3βΔ9, and then maintained in low-serum medium for 16 h. 2D-PAGE immunoblots were revealed against anti-V5 antibody (NURR1). Black arrows indicate acidic spots that result from GSK-3 phosphorylation and are lost in NURR1MUT2-V5. C, half-life of NURR1MUT2-V5 is not affected by GSK-3β. HEK293T cells were co-transfected with NURR1-V5, NURR1MUT2-V5 together with HA-GSK-3βY216F or HA-GSK-3βΔ9, serum starved for 16 h, and finally incubated for the indicated time with 100 µM CHX. Upper blot, anti-V5 (NURR1WT-V5 or NURR1MUT2-V5) protein levels. Middle blot, HA-GSK-3β mutants. Lower blot, GAPDH levels showing similar protein loads per lane. D, the graph depicts the natural logarithm of the relative levels of the NURR1WT-V5 and NURR1MUT2-V5 protein as a function of CHX incubation time. The protein half-life was determined using the linear part of the degradation curve. Statistical analysis was performed with one-way ANOVA followed by Newman–Keuls multiple comparison test. ***p < 0.001. E Ubiquitilation of NURR1MUT2-V5 is drastically reduced despite the presence of active GSK-3β. HEK293T cells were co-transfected with the indicated plasmids or without HA-Ub vector (Ub) as control. One fifth of whole-protein lysate was used to control for protein expression as shown in the three upper panels (total Input). The rest of the protein lysates were immunoprecipitated with anti-V5 antibody and immunoblotted as anti-HA to detect ubiquitinated NURR1
To more precisely characterize the relevance of Core 2, HEK293T cells were transfected with NURR1WT or NURR1MUT2 and co-transfected with GSK-3βΔ9 or GSK-3βY216F, and resolved by 2D gel electrophoresis. As shown in Fig. 6B, NURR1WT co-transfected with hypomorphic GSK-3βY216F, displayed several immunoreactive spots consistent with GSK-3β-independent posttranslational modifications of NURR1. When cells were co-transfected with active GSK-3βΔ9, we observed an increase in the intensity of acidic spots (black arrows), indicating GSK-3-mediated phosphorylation. However, in cells co-transfected with NURR1MUT2 the distribution of spots was similar in the presence of GSK-3βY216F or GSK-3βΔ9, and the acidic shift was not observed (black arrows). These results indicate that the residues of Core 2 are preferred targets of GSK-3β phosphorylation.
In additional experiments, we examined the half-life of NURR1MUT2. HEK293T cells were co-transfected with NURR1WT or NURR1MUT2 and GSK-3βΔ9, and exposed to CHX (100 µM) (Fig. 6C, D). In the presence of GSK-3βΔ9, the half-life of NURR1WT was less than 120 min. However, NURR1MUT2 exhibited a half-life of more than 120 min that was not significantly shortened by the presence of GSK-3βΔ9. We also performed an ubiquitination assay in HEK293T cells co-transfected with expression vectors for NURR1WT or NURR1MUT2 along with HA-Ubiquitin and GSK-3βY216F or GSK-3βΔ9. As shown in Fig. 6E, overexpression of GSK-3βΔ9 enhanced NURR1WT ubiquitination, while in NURR1MUT2 this was less apparent.
Finally, we validated the phosphorylation of Core 2 in the DAergic cell line SH-SY5Y (Fig. 7A). These cells were infected with lentiviral vectors expressing NURR1WT or NURR1MUT2. In the presence of DOX, α-SYN levels were low in cells expressing either form of NURR1, and the levels of GSK-3β-pSer9 were high, indicating its inhibition. However, in the absence of DOX, α-SYN levels increased, and GSK-3 was dephosphorylated and active. The presence of α-SYN and its downstream target, active GSK-3, completely eliminated NURR1WT protein and substantially reduced the levels of TH and RET. By contrast, the levels of NURR1MUT2 were not affected by the α-SYN/GSK-3 challenge and the levels of TH and RET remained similar to those in the absence of α-SYN.

GSK-3β activation by conditional expression of α-SYN or by phosphatidylinositol-3 kinase (PI3K) inhibition leads to loss of NURR1 stability. A SH-SY5Y cells carrying the α-SYN Tet-Off expression system were treated with or without 2 µg/ml DOX for 5 days in the presence RPMI with serum. Then, the cells were infected with lentiviruses expressing to NURR1WT-V5 and NURR1MUT2-V5. After 24 h, cells were transferred to Opti-MEM Reduced Serum Medium maintaining the DOX treatment for 8 days. B SH-SY5Y cells were maintained in Opti-MEM Reduced Serum Media for 16 h and then pre-treated with SB216763 (5 μM, 2 h) prior to inhibition of the PI3K/AKT pathway with 30 µM LY294002. β-Catenin protein levels were used as positive control GSK-3 activity. AKT-pSer473 and GSK-3β-pSer9 levels were used as control LY294002 treatment. Finally, GAPDH protein levels show similar protein loaded per lane. C Densitometry quantification of NURR1 protein levels normalized with GAPDH from representative blots of cells treated like in B. Each value is the mean ± S.E.M. (n = 3). Statistical analysis was performed with one-way ANOVA followed by Newman–Keuls multiple comparison test. ***p < 0.001
A highly characterized survival pathway in nerve cells is the PI3K/AKT. This pathway is activated by many growth factors and neurotrophins and leads to the inhibition of GSK-3. Therefore, we analyzed the inhibition of this pathway, by using the highly selective PI3K inhibitor LY294002. SH-SY5Y cells were submitted to a time-course of LY294002 (30 µM) alone or in combination with the GSK-3 inhibitor SB216763 (5 μM). As shown in Fig. 7B, C, LY294002 alone led to a decrease in AKT-pSer473 (inactivation) and GSK-3β-pSer9 (activation). Under these conditions, not only β-catenin but also NURR1 levels were gradually decreased. By contrast, cells co-treated with SB216763 were at least partially protected from the decrease in β-catenin and NURR1. Together, these observations confirm in a DAergic cell line the mechanistic connections between α-SYN, GSK-3β, phosphorylation, and degradation of NURR1.
