
Abstract
Mechanical ventilation with large tidal volumes can increase lung alveolar permeability and initiate inflammatory responses, termed ventilator-induced lung injury (VILI). VILI is characterized by an influx of inflammatory cells, increased pulmonary permeability, and endothelial and epithelial cell death. But the underlying molecular mechanisms that regulate VILI remain unclear. The purpose of this study was to investigate the mechanisms that regulate pulmonary endothelial barrier in an animal model of VILI. These data suggest that SC5b-9, as the production of the complement activation, causes increase in rat pulmonary microvascular permeability by inducing activation of RhoA and subsequent phosphorylation of myosin light chain and contraction of endothelial cells, resulting in gap formation. In general, the complement-mediated increase in pulmonary microvascular permeability may participate in VILI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High tidal volume mechanical ventilation (HVT MV) during critical care can cause structural and functional disturbances in the lung with subsequent release of proinflammatory mediators, that is, ventilator-induced lung injury (VILI) [1]. The pathologic effects of VILI include destruction of both capillary endothelium and alveolar epithelium, alveolar flooding, lung nonuniformity, terminal airway closing and reopening, neutrophils recruitment, and the release of inflammatory mediators [2–6]. Although it has been studied extensively for the past several decades, the accurate mechanisms of VILI are still not well known. In early studies, the development of pulmonary edema during mechanical ventilation was ascribed to increased lung microvascular pressure, resulting from high lung volume ventilation and surfactant depletion [7]. However, the magnitude of microvascular pressure changes during lung inflation is modest and insufficient to explain the occurrence of severe pulmonary edema during mechanical ventilation with a high tidal volume. In addition to pressure changes, alterations of alveolar–capillary barrier permeability are involved in the development of pulmonary edema during high-volume ventilation of intact animals and are the most important responsible for VILI [8].
It is now clear that ventilation-induced pulmonary edema is essentially the result of severe changes in the permeability of the alveolar–capillary barrier; disruption of the alveolar–capillary barrier is an important mechanism responsible for the formation of alveolar edema, which is characteristic of VILI [9]. In a sentinel study, Webb and Tierney reported that high tidal volume ventilation-induced pulmonary edema and diffuse alveolar damage histologically indistinguishable from ARDS in a rat model. It was demonstrated that, within minutes after the onset of ventilation, rats ventilated with 45 cm H2O peak pressures exhibited not only macroscopic pulmonary edema but also a dramatic increase in microvascular permeability assessed by the distribution space of intravenously injected 125I-labeled albumin [10]. Electron microscope observations clearly revealed widespread disruption of epithelial cells leading to denudation of basement membranes and the presence of many gaps in the capillary endothelium [11]. Such findings demonstrated that high-volume mechanical ventilation leads to pulmonary edema of the permeability type. These electron microscopy findings strongly support the contention that permeability alterations are a main determinant of ventilator-induced pulmonary edema.
Ventilator-induced lung injury also has typical inflammation changes, which is supported primarily by observations made in animals at high-volume mechanical ventilation and suggests that inflammation plays vital role in the mechanisms of VILI [12]. The biotrauma hypothesis, that is, lung tissue stretching might result in lung damage solely through the release of inflammatory mediators and leukocyte recruitment, has been put forward to provide an explanation why most patients with ARDS die from multiple organ failure rather than hypoxemia [13].
The complement cascade, a major component of innate immunity, represents a driving force in the pathophysiology of multiple inflammatory disorders [14–16]. Studies have shown that several products formed in the complement pathway participate in the complement-mediated microvascular injury [17]. A recent study has demonstrated that complement 3 is involved with mechanisms causing VILI [18]. Clinical observations suggested association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome [19]. These observations support that complement activation could mediate VILI. While the precise role of complement in VILI remains undetermined. Since blood levels of vitronectin-containing complement complexes SC5b-9 increase in ARDS [20], the ligation of the endothelial luminal αvβ3 integrin by this soluble terminal complement complex (SC5b-9) has been proved to increase lung endothelial liquid conductance [21]; we conclude that complement activation may be a potential cause of the lung microvascular injury that underlies pulmonary edema of conditions such as VILI. However, specific SC5b-9-mediated signaling events that affect endothelial barrier properties remain poorly understood. The present study was designed to evaluate the role of complement complex SC5b-9 levels, the extent of complement activation, and of complement hemolytic activity in serum, as potential new contributor for the severity of VILI.
Though permeability alterations are obvious and severe during ventilator-induced edema, the underlying mechanisms are not fully understood. It has been well documented that endothelial barrier function is controlled by the stability of intercellular junctions as well as tensile force within the monolayer maintained by the actomyosin cytoskeleton. Contractile forces generated by actomyosin interaction tend to pull the tightly connected cells apart [22]. The physical process of “pulling” endothelial cells apart is a downstream event in multiple signaling pathways activated by inflammatory mediators [23–27], such as those generated by VILI. Rho family GTPases regulated both actin cytoskeletal organization and the integrity of intercellular junctions and have been implicated in intracellular signaling induced by many vasoactive substances including thrombin, tumor necrosis factor alpha (TNF-α), bradykinin, histamine, lysophosphatidic acid (LPA), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF) [28–33]. Although previous experiments showed that SC5b-9 caused a hydraulic conductivity (Lp) increase through a signaling pathway involving tyrosine kinase activation [21], there are no direct evidence proved that SC5b-9 can activate Rho A/ROCK signaling pathway.
The purpose of this study was to assess the contribution of complement activation to pulmonary microvascular dysfunction elicited by HVT MV. With the use of an in vitro endothelial barrier model, we found that application of SC5b-9 to rat pulmonary microvascular endothelial cell (RPMVEC) monolayers resulted in increases in permeability and myosin light chain (MLC) phosphorylation. This hyperpermeability and MLC phosphorylation was shown to be dependent on Rho A/ROCK activation. Furthermore, we showed increased F-actin levels and alterations in the cytoskeleton marked by stress fiber formation upon treatment of the cells with SC5b-9.
Materials and Methods
Reagents
Culture medium M-199, colustrum-free bovine serum, antibiotic–antimycotic mixture, and nonessential amino acids were obtained from GIBCO BRL (Chagrin Falls, OH, USA). Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO, USA). FITC-phalloidin was purchased form Molecular Probes (Eugene, OR, USA). phospho-MLC antibody was obtained from Cell Signaling (Beverly, MA, USA); G-LISA™ RhoA activation assay biochemistry kit was obtained from Cytoskeleton (Denver, CO, USA).
Rat Model of VILI
To induce the VILI, adult male Sprague–Dawley rats weighing 250–300 g were maintained on a 12-h light–dark cycle with free access to food and water. Rats were intraperitoneally anesthetized with pentobarbital (50 mg/kg). A tracheostomy was performed, and a 14-gauge plastic cannula was inserted into the trachea. Animals were then subjected to mechanical ventilator (Model683, Harvard Apparatus, Boston, USA) at a VT of 40 mL/kg, zero positive end-expiratory pressure (PEEP), a respiratory rate of 60 breaths/min, and Fi O2 of 0.21 for 0 h (sham, n = 6), 2 h (VT-2, n = 6), or 4 h (VT-4, n = 6). Sham-operated animals underwent the same surgical procedures [34]. Another group that received no ventilation served as the control group (Ctrl, n = 6). All animals were kept supine for the duration of the experiment.
Plasma Complement Activity
Total hemolytic complement activity was determined by an in vitro liposome immunoassay (LIA) with the Autokit CH50, a commercial liposome immunoassay (Wako, Osaka, Japan) in an automated analyzer. Serum levels of CH50 were determined on serum samples frozen immediately after extraction at −20 °C and subsequently stored at −80 °C. Repetitive freeze–thaw cycles were avoided to minimize in vitro complement activation.
Evans Blue Dye Assay
To evaluate permeability of the rat lung to macromolecules after exposed to HVT MV. Evans blue dye (EBD) was dissolved in 0.9 % saline at a final concentration of 5 mg/mL. Animals were anesthetized, weighed, and injected with 20 mg/kg EBD in the left femoral vein. After 30 min, the animals were killed and the lungs perfused with 1 mL PBS containing 5 mM ethylenediaminetetraacetic acid. The lungs were excised en bloc and snap-frozen in liquid nitrogen. The frozen lungs were homogenized in 2 mL PBS. The homogenate was diluted with 2 vol of formamide and incubated at 60 °C for 2 h, followed by centrifugation at 5,000×g for 30 min. The supernatant was collected, and absorbance was measured at 620 and 740 nm in a dual-wave spectrophotometer [35, 36]. The EBD concentration was determined from standard absorbance curves evaluated in parallel. Correction for contaminating heme pigments was calculated by the formula E620 (EBD) = E620 − (1.426 × E740 + 0.030). The EBD concentration was expressed as a percentage of the total dose of EBD administered [37].
Preparation of SC5b-9
SC5b-9 was purified by the modified method of Tsukada et al. [21], with some modifications as described. Briefly, SC5b-9 was precipitated from zymosan-activated serum (ZAS) (9 mL) by the addition of ammonium sulfate to a 37.5 % concentration while the mixture was stirred in an ice bath. The precipitate was recovered by centrifugation, dissolved in PBS (pH 7.4, 2 mL) containing 1 % Triton X-100, and immediately chromatographed on a column of Sepharose CL-6B equilibrated with PBS. Fractions were electrophoresed on cellulose acetate and stained (with amido black) to identify proteins. Some samples were Western blotted with anti-vitronectin antibody to confirm the presence of vitronectin in SC5b-9. SC5b-9 levels in the fractions were quantified by BCA using a microplate spectrophotometer (Biotek, Sunnyvale, CA, USA).
Cell Cultures
Rat pulmonary microvascular endothelial cells (RPMVECs) were isolated using a modification of a technique previously described by Chen et al. [38]. Pathogen-free male Sprague–Dawley rats, 120–150 g were anesthetized with ketamine hydrochloride (100 mg/kg im) and heparinized (1,000 units per animal) intraperitoneally. After sedation, the animals were secured in the supine position, and the chest and upper abdomen were shaved, scrubbed with betadine, and washed with 75 % ethanol. With sterile technique, the chest cavity was opened, and the animal was killed by exsanguination. The lungs were removed and placed in a beaker containing Hanks’ buffered salt solution (HBSS) with 2 % antibiotic–antimycotic solution. The visceral pleura were first stripped from each lobe, and the outer 3- to 5-mm of the peripheral lung tissue were sharply dissected free from the remaining tissue and were cut into pieces of 1 × 1 × 1.5 mm, separately. Five pieces were placed into a 35-mm dish gelatinized with 0.5 % gelatin and cultured in M199 media supplemented with 20 % (v/v) colostrum-free fetal bovine serum. The plates were placed in a humidified incubator at 37 °C with 5 % CO2. After 60 h of culture, the tissues were discarded and the medium was partially changed. The dish contained only endothelial cells and blood cells. Blood cells were removed by subculture. The endothelial cells were subcultured with 0.08 % trypsin in D-Hanks’ solution between the sixth and tenth day. The subcultured cells grew to contact-inhibited monolayers with the typical cobblestone morphology. The cells were maintained for experimental use.
Endothelial Monolayer Permeability Assay (TEER)
RPMVECs were passaged when they reached 80 % confluency, using 0.08 % trypsin in EDTA. After washing twice with D-Hanks’ solution, cells were seeded on a gelatin-coated, 24-well transwell polycarbonate membrane tissue culture dish (6.5 mm diameter, 3.0 μm pore size, Corning Costar, Tewksbury, MA, USA) at a density of 1 × 105 cells per well and placed into lower chamber containing 600 μL of complete medium. RPMVECs were used after 24 h of culture to attain monolayer adhered on the transwell polycarbonate membrane. Transendothelial electrical resistance (TEER) across RPMVECs was measured using Millicell-ERS (Millipore, Billerica, MA, USA) at various incubation time (1, 2, 3, 4 h) after addition of either vehicle (M199) or agonist (SC5b-9) at indicated concentration (μg/mL) into the luminal chamber (n = 3 per group). The TEER value was first measured at 24 h after seeding, and this time point was defined as 0 h. Every test was done in duplicate and repeated at least three times. The upper and lower chamber media were not changed until the end of experiment. The TEER value was calculated by the formula: [the average resistance of experimental wells − the average resistance of blank wells] × 0.33 (the area of the transwell membrane) [39, 40].
Immunofluorescent Staining
RPMVECs grown on glass coverslips were fixed after either vehicle (M199) or agonist (SC5b-9) treatment in 3.7 % formaldehyde solution in PBS for 10 min at +4 °C, washed three times with PBS, permeabilized with 0.2 % Triton X-100 in PBS-Tween (PBST) for 30 min at room temperature, and blocked with 2 % BSA in PBST for 30 min. Actin filaments were stained with FITC-conjugated phalloidin for 1 h at room temperature. After immunostaining, the glass slides were analyzed using an Olympus BX51 microscope (Olympus, Shinjuku-ku, Tokyo) connected to digital camera and image processor. The images were recorded and processed using Adobe Photoshop 6.0 program.
Image Analysis of Stress Fiber and Intercellular Gap Formation
FITC-phalloidin-stained EC monolayers stimulated with either SC5b-9 or vehicle were viewed at 1,000× magnification under fluorescence microscope and recorded as described above. The images were analyzed using Image-Pro 5.02 image processing program (Media Cybernetics, USA). Images were differentially segmented between gaps (black) and cells (highest gray value) based on image grayscale levels. The intercellular gaps were also analyzed with Image-Pro 5.02 to obtain the area of each gap, and the data are presented as the percentage of area taken up by intercellular gaps relative to the total area of the field examined. The values were statistically processed using Excel (Microsoft, Redmond, WA, USA) software.
Western Blot
RPMVECs were seeded (2 × 105 cells) into six-well plastic culture plates and incubated for 3–4 days until confluent (1.2 × 106 cells). EC monolayers were stimulated with either vehicle (M199) or SC5b-9 for the indicated times (1, 2, 3, 4 h) at a concentration of 150 μg/mL, which was chosen due to the fact that a higher concentration of SC5b-9 markedly interfered with the integrity of the monolayer and the TEER across the Transwell inserts. After interventions, the RPMVECs were washed with ice-cold PBS, lysed in 200 μL extraction buffer (20 mM Tris–HCl (pH 7.4), and collected in microtubes. After centrifugation (16,000×g, 20 min, +4 °C), protein extracts (10–12 mg) solubilized in 3 × SDS sample buffer and were separated on 10 % SDS–polyacrylamide gels and electroblotted to a nitrocellulose membrane. Phosphorylated MLC were detected by phospho-MLC antibody; β-actin was used as internal control to correct for variations of different samples. Protein bands were visualized by a G- BOX detection system and quantified determining the change in integrated optical intensity (IOI) using the GeneTools analysis software (Syngene, Cambridge, UK).
RhoA GTPase Activation Assay
Rho activation in RPMVEC culture was analyzed using the luminescence-based G-LISA RhoA activation assay biochemistry kit according to the manufacturer’s instructions. Briefly, RPMVECs were grown to confluence. The cells were challenged with or without SC5b-9 (150 μg/mL) for the indicated times (1, 2, 3, 4 h). At the end of experiment, cells were rinsed with ice-cold PBS. Protein was isolated using the provided cell lysis buffer, and cells lysates were processed rapidly on ice and snap-frozen and stored at −80 °C until the time of assaying. Cell lysates were subsequently thawed and clarified by centrifugation at 14,000 rpm at +4 °C for 2 min. Protein concentration was determined according to the manufacturer’s protocol, and cell extracts were equalized to contain protein concentrations of 2 mg/mL for the assay. Equal total protein amounts were added to a 96-well dish coated with the Rho-binding domain of Rho effector proteins (which bind active GTP-bound Rho) in duplicate and incubated at +4 °C for 30 min with vigorous shaking. Active Rho levels were determined by subsequent incubations with anti-Rho antibody and secondary horseradish peroxidase-conjugated antibody for 45 min each with vigorous shaking at room temperature. After adding develo** solution, active Rho was determined by detecting the luminescence signals at 490 nm using an FlexStation II microplate luminescence reader (Molecular Device, Sunnyvale, USA) after subtraction from a sample containing only lysis buffer [41].
Statistical Analysis
For permeability experiments, ONE WAY ANOVA was performed to determine the rate of EBA diffusion in individual wells with Excel 2003 software. The means and SD were determined from these slopes. For three or more groups, differences among the means were tested for significance in all experiments, using ANOVA with Fisher’s least-significance-difference test. Significance was reached when P < 0.05. All data are presented as mean ± SD. The n is indicated for each set of data.
Results
Complement Levels and Activity in the Normal and HVT MV-Injured Rat
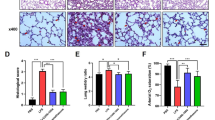
The complement system plays an important role in host defense against infection and in most inflammatory processes. The standard 50 % hemolytic complement (CH50) assay is the most commonly used method of screening patient sera for functional activity of the classical complement pathway. CH50 tests showed that the serum complement activity in animals subjected to mechanical ventilator was more significantly higher (P < 0.05) than that of the control or sham group. In this experiment, elevation of the CH50 in both the VT-2 and VT-4 groups confirmed those results (Fig. 1).

Effect of HVT MV on systemic complement activity in rats. Percent changes in 50 % hemolytic unit of complement serum (CH50) of the rat sera was increased at 2 and 4 h of HVT MV compared with control and Sham animals. Significantly different, *P < 0.05 versus control group. Results are mean ± SD from 5 animals in each group
Lung Microvascular Leak Measured by Evans Blue Dye
EBD leak in the lung was significantly higher in HVT MV group compared with the control or sham group (Fig. 2). There was a fivefold increase in the lung/total dose ratio of EBD after 4 h of HVT MV exposure. These data are consistent with the histology demonstrating that lung edema was significantly induced by mechanical ventilation.

Extravasation of Evans blue dye (EBD) into the rat lung tissue after HVT MV. Lung tissues were homogenized in formamide and extracted overnight, and EBD concentration was determined as described in “Materials and Methods” section. Results are expressed as total EBD/lung. *P < 0.05 results are mean ± SD from 6 animals in each group. Hyperpermeability in mechanically ventilated rat lungs were identified by Evans Blue Assay (**P < 0.01)
Effect of SC5b-9 on Pulmonary Endothelium Barrier Properties
Measurements of TEER across pulmonary EC monolayers after SC5b-9 stimulation demonstrated that SC5b-9 significantly decrease TEER of EC monolayer reflecting dramatic EC barrier dysfunction. Figure 3 shows the transendothelial electrical resistance across EC monolayer that were treated with either vehicle or SC5b-9 (50, 100, 150, 200 μg/mL, respectively) for indicated times (1, 2, 3, 4 h) reveal patterns of time- and dose-dependence. Maximal TEER decrease was achieved at 4 h of stimulation with 200 μg/mL SC5b-9.

Effect of SC5b-9 on transepithelial electrical resistance (TEER) in insert-cultured rat pulmonary microvascular endothelial cells. Confluent cells were stimulated with either M199 (vehicle control) or SC5b-9 (50, 100, 150, 200 μg/mL, respectively) for indicated time(1, 2, 3, 4 h). SC5b-9 promote a low TEER in EC monolayers. Data are expressed as mean ± SD (n = 3). (Asterisk) M199; (filled square) 50 μg/mL SC5b-9; (filled triangle) 100 μg/mL SC5b-9; (filled diamond) 150 μg/mL SC5b-9; (open circle) 200 μg/mL SC5b-9
SC5b-9-Induced Cytoskeletal Changes in Pulmonary Endothelium
Because cellular contraction and gap formation are linked to formation of stress fibers, we looked at actin distribution in response to SC5b-9 stimulation. Immunofluorescent staining using FITC-phalloidin demonstrated that SC5b-9-induced decreases in TEER were linked to actomyosin rearrangement. In unstimulated cells, F-actin primarily organized into cortical band lining the cell cortex peripherally (Fig. 4, left panel, short arrows). After SC5b-9 stimulation, F-actin reorganized into shorter, thicker stress fibers in the center of the cells (Fig. 4, right panel and long arrows), whereas peripheral actin band disappeared. These changes were associated with appearance of paracellular gaps, indicating contraction of endothelial cells (Fig. 4, right panel). Stress fiber formation persisted corresponding to the time point of TEER decrease.

Effect of SC5b-9 on endothelial cytoskeletal arrangement. Confluent ECs grown on glass coverslips were challenged either with vehicle (M199) or SC5b-9 (150 μg/mL) for the indicated periods of time. After stimulation, cells were fixed and stained with FITC-phalloidin for F-actin. SC5b-9 caused significant stress fiber formation and development of intracellular gaps (shown by long arrows in the right panel) accompanied by disappearance of cortical ring (shown by short arrows in the left panel), disruption of cell–cell contacts. Shown are representative results of three independent experiments
SC5b-9-Induced Gap Formation in Pulmonary Endothelium
Previous studies have demonstrated that agonist induced increases in pulmonary EC permeability are associated with appearance of paracellular gaps [42]. To find whether SC5b-9-induced hyperpermeability also associated with gap formation, we performed Immunofluorescent staining using FITC-phalloidin (Fig. 5, Panel a–e). Quantitative analysis (Fig. 5, Panel f) confirmed increased gap formation indicative of cell contraction observed after SC5b-9 stimulation.

Effect of SC5b-9 on endothelial monolayer integrity. Confluent ECs grown on glass coverslips were challenged either with vehicle (M199) or SC5b-9 (150 μg/mL) for the indicated periods of time. After stimulation, cells were fixed and stained with FITC-phalloidin for F-actin. Gap formation induced by SC5b-9 was assessed by morphometric analysis of phalloidin-stained RPMVEC performed using Image-Pro 5.02 image processing software as described in “Materials and Methods” section. Gap formation was assessed by increases in the ratio of the integrated gap area to the area of the whole image. Shown are representative results of three independent experiments
Effect of SC5b-9 on RPMVECs MLC Phosphorylation
Phosphorylation of MLC by activated myosin light chain kinase (MLCK) plays a critical role in the development and regulation of contractile forces within cells [43]. Bold studies have shown that the phosphorylation condition of MLC affects the permeability of cultured endothelial cells. The activation of MLC is marked by increases in serine/threonine phosphorylation. Inhibition of serine phosphorylation attenuates agonists-induced MLC phosphorylation and decreases permeability across endothelial cells [44]. As Fig. 6 shows, phosphorylation of MLC increased dramatically after 3 h exposure to SC5b-9. In SC5b-9 treated for 4 h group, the level of MLC phosphorylation increased to 1.5 of that of the control group (compare lanes 4h and Veh). Inhibition of MLCK using pharmacological inhibitor ML-7 attenuates SC5b-9-induced MLC phosphorylation and decreases permeability across endothelial cells. These findings suggest that both MLCK activation and MLC phosphorylation are required for HVT MV induced lung injury.

Effect of SC5b-9 on EC myosin light chain (MLC) phosphorylation. Confluent ECs were treated with either vehicle (M199) or SC5b-9 for indicated periods of time. MLC phosphorylation was determined as described in “Materials and Methods” section. Each bar represents the mean ± SD of 3 independent experiments.*P < 0.05; **P < 0.01
Effects of SC5b-9 Stimulation on RhoA Activity
The small GTPase RhoA is known to be a critical regulator of actin stress fiber formation as well as an important regulator of increased endothelial permeability. Having shown that phosphorylation status of MLC is essential for hyperpermeability in response to SC5b-9, we sought to examine whether SC5b-9-induced permeability changes are regulated at the level of RhoA activation. We used a luminescence-based G-LISA assay to assess global SC5b-9-induced increases in cellular RhoA activity. Values were obtained from three separate experiments performed in triplicates and are represented as mean ± SD (P < 0.05, SC5b-9 vs. control). We found robust RhoA activation by SC5b-9 in RPMVECs (Fig. 7). SC5b-9-induced changes in endothelial permeability correlate with changes in the activity of RhoA. This finding suggests that RhoA activation contributes to SC5b-9 mediated increase in endothelial permeability. As shown in Fig. 7, when cells were exposed to the Rho kinase (ROCK) inhibitor Y-27632 before exposure to SC5b-9, the permeability response was greatly attenuated. Together with the permeability experiments using MLCK inhibitors, the data strongly suggest that both MLCK and Rho kinase are essential for SC5b-9 induced hyperpermeability and MLC phosphorylation.

SC5b-9 treatment causes RhoA activation. RPMVECs were subjected to SC5b-9 for the times indicated. The active form of RhoA was luminometrically detected using a G-LISA kit. GTP-loading assays of Rho were performed as described in “Materials and Methods” section. RhoA activity increased significantly in the SC5b-9-stimulated cells. Values are mean ± SD from 3 independent experiments. *P < 0.05; **P < 0.01, comparisons with M199 control. RPMVECs grown on transwell inserts were treated with Y-27632 (5 μM) for 30 min before being exposed to SC5b-9 at 150 μg/mL concentration for the times shown. TEER across EC monolayer was recorded for indicated time (0, 1, 2, 3, 4 h)
Discussion
Mechanical ventilation, although a lifesaving intervention in the care of the critically ill patient, can result in physical trauma to lung tissue (VILI). In the last several decades, numerous studies have provided evidence that lung injury is often associated with mechanical ventilation. This ventilator-induced lung injury may impair the function of healthy lungs, or exacerbate injury in the diseased lung (i.e., due to infection or trauma). It was known that the acute pulmonary injury secondary to mechanical ventilation is complex and multifactorial [45]. Although little doubt exists that mechanical ventilation (especially at high volume) can physically disrupt the lung, recent studies have shown that mechanical ventilation also results in more subtle morphological and functional changes and can excite an inflammatory response within the lung, a mechanism called “biotrauma” [46]. Indeed, several studies have found that mechanical ventilation is associated with a systemic inflammatory response [47–50].
The complement system, a set of more than 35 proteins in plasma, is an effector of several functions associated with inflammation, immunologic regulation, and cytotoxicity [51]. The importance of complement system in the development and amplification of the inflammatory process at the tissue level in various pathological conditions has been previously demonstrated. Complement activation products contribute to a large number of inflammatory diseases [52]. There is evidence that suggests the activation of the complement cascade contributes to the pulmonary microvascular dysfunction observed in ARDS [53]. For example, complement-activated serum increases lung microvascular Lp and that the critical factor responsible is SC5b-9 [54]. Therefore, the link between complement activation and the endothelial barrier dysfunction may be attributable to the formation of the complement complex SC5b-9 in the terminal part of the complement pathway. SC5b-9 is known to contain several molecules of the prototypic αvß3 ligand vitronectin, which recognizes the integrin by means of its RGD (Arg-Gly-Asp) tripeptide sequence. As a classic αvß3 ligand in the native monomeric state, vitronectin has no detectable effect on the lung endothelial barrier, but several vitronectin molecules are incorporated into SC5b-9 following complement activation [55]. The juxtaposition of multiple vitronectin molecules, as in SC5b-9 is likely to increase the density of RGD sites, which may lead to ligation of multiple αvß3 receptors, receptor aggregation, and the induction of signal transduction mechanisms. The cytolytically inactive soluble terminal complement complex SC5b-9 exerts several pro-inflammatory responses acting directly on endothelium, such as the induction of adhesion molecules and the increase in vascular leakage [56, 57]. In this study, detection of complement level and pulmonary vascular permeability in VILI rats showed that the complement levels in HVT MV-exposed rats were significantly elevated (P < 0.05). High tidal volume mechanical ventilation appears to stimulate complement activation, which confirm the importance of complement activation in mediating lung injury after HVT MV. Hyperpermeability in HVT MV-exposed rat lungs were identified by Evans Blue Assay (P < 0.01). These results provide a mechanism to injure the endothelial barrier, thereby allowing passage of protein-rich fluid into the interstitial and alveolar spaces.
Mechanical deformations during high airway pressure ventilation may produce various cellular effects. Tissue stretch may result in direct damage (the extreme limit being ruptures as during the classical barotrauma). More subtle events may also occur, including the triggering of inter- and intracellular signals [58]. This suggests that the increase in microvascular permeability may not be a simply passive physical phenomenon, but the result of biochemical reactions.
As stated in the introduction, endothelial barrier function is maintained in a semipermeable state by the balance between adhesive forces at intercellular junctions and contractile forces, which are produced by the actomyosin complex, within the cells. The complement levels in HVT MV-exposed rats were significantly elevated which suggest that complement complex SC5b-9 stimulation might play a role in the process of endothelial cell retraction. Immunofluorescent staining using FITC-phalloidin demonstrated that actomyosin rearrangement was linked to SC5b-9-induced paracellular gap formation, indicating contraction of endothelial cells. Stress fiber formation persisted corresponding to the time point of TEER decrease. Exposure of rat pulmonary microvascular endothelial cells to activated complement product SC5b-9 results in significant endothelial hyperpermeability associated with cytoskeletal reorganization. These results suggest that the reorganization of the endothelials cytoskeleton and net filamentous actin (F-actin) assembly stimulated by SC5b-9 contributes to complement-mediated endothelial barrier dysfunction.
Although the mechanisms of SC5b-9-induced EC barrier dysfunction involving tyrosine kinase activation have been described by others [59], the detailed role of Rho-mediated signaling in SC5b-9-induced EC barrier regulation has not been investigated. We are now able to study the regulatory mechanisms at the molecular level using a monolayer permeability system. Endothelial cells derived from rat pulmonary microcirculation were used to perform detailed analysis of Rho-mediated pathway involved in SC5b-9-induced EC barrier dysfunction. This study, for the first time, linked complement to development of pulmonary microvascular endothelial cell hyperpermeability in an MLC phosphorylation-dependent manner. With the knowledge that the small GTPase Rho A is a critical regulator of actin stress fiber formation as well as an important regulator of increased endothelial permeability, we sought to determine whether Rho A activity change is involved in SC5b-9-induced barrier dysfunction. Data acquired with a luminescence-based G-LISA assay suggests that Rho A activation contributes to SC5b-9-mediated induction of stress fiber formation and increased endothelial permeability. However, there is a discrepancy between SC5b-9-induced changes in endothelial permeability and the activity of Rho A. Peak Rho A activity attained 1 h after SC5b-9 stimulation, then declined with time, while TEER in the SC5b-9-stimulated cells decreased linearly. The reason of this discrepancy is that Rho A activity varies quickly in a ligand-dependent manner under different context within the cell [60], while SC5b-9 stimulation could weaken the intercellular junctions which will lead to prolonged TEER decline. Furthermore, inactivation of Rho kinase (ROCK) greatly attenuated the permeability response to SC5b-9 challenge. Taken together, SC5b-9 could induce endothelial hyperpermeability by activating Rho A/ROCK signaling pathway.
In this study, we established an in vitro model of SC5b-9-induced endothelial monolayer permeability dysfunction. Endothelial permeability was significantly increased by SC5b-9 challenge in a time- and concentration-dependent manner and was accompanied by changes in the F-actin cytoskeleton. The present model of SC5b-9-elicited lung injury is also unique in that it demonstrates this soluble complement complex is a potent inflammatory stimulus that disrupts normal lung microvascular integrity. In the utilization of this model, the signal of complement-induced microvascular injury in the lung can be individually examined. The fact that enhanced lung microvascular barrier dysfunction is attenuated by inhibition of cytoskeletal regulating cascades before SC5b-9 stimulation provides direct evidence that complement activation in the lung is a critical component of the systemic inflammatory response syndrome. These results confirm the importance of complement activation in mediating lung injury after HVT MV.
In conclusion, our results showed that HVT MV could activate complement system, leading to the formation of vitronectin-containing complement complex SC5b-9. The stimulation of the endothelial cells by these complexes induces cytoskeletal reorganization and further evokes an increase in vascular permeability. Activation of the Rho A/ROCK signal transduction pathway appears to be an important mediator of SC5b-9-mediated pulmonary dysfunction elicited by HVT MV. The involvement of Rho A/ROCK signaling pathway and complement activation provides a new area for finding therapeutic measure for ventilator-induced lung injury. The results of the present investigation are unique in that it provides direct evidence that complement complex-mediated increases in vascular hyperpermeability occur after mechanical ventilation in vivo. We propose that SC5b-9 is involved with VILI and inhibition of complement activation may be a potential therapeutic strategy to limit injuries in mechanically ventilated patients.
References
Boost, K. A., Hoegl, S., Dolfen, A., Czerwonka, H., Scheiermann, P., Zwissler, B., et al. (2008). Inhaled levosimendan reduces mortality and release of proinflammatory mediators in a rat model of experimental ventilator-induced lung injury. Critical Care Medicine, 36, 1873–1879.
Dreyfuss, D., Basset, G., Soler, P., & Saumon, G. (1985). Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. The American Review of Respiratory Disease, 132, 880–884.
Slutsky, A. S. (1999). Lung injury caused by mechanical ventilation. Chest, 116, 9S–15S.
Woo, S. W., & Hedley-Whyte, J. (1972). Macrophage accumulation and pulmonary edema due to thoracotomy and lung overinflation. Journal of Applied Physiology, 33, 14–21.
Narimanbekov, I. O., & Rozycki, H. J. (1995). Effect of Il-1 blockade on inflammatory manifestations of acute ventilator-induced lung injury in a rabbit model. Experimental Lung Research, 21, 239–254.
Vlahakis, N. E., Schroeder, M. A., Limper, A. H., & Hubmayr, R. D. (1999). Stretch induces cytokine release by alveolar epithelial cells in vitro. American Journal of Physiology, 277, L167–L173.
Parker, J. C., Hernandez, L. A., Longenecker, G. L., Peevy, K., & Johnson, W. (1990). Lung edema caused by high peak inspiratory pressures in dogs. Role of increased microvascular filtration pressure and permeability. The American Review of Respiratory Disease, 142, 321–328.
de Prost, N., Ricard, J. D., Saumon, G., & Dreyfuss, D. (2011). Ventilator-induced lung injury: Historical perspectives and clinical implications. Annals of Intensive Care, 1, 28–43.
Dreyfuss, D., & Saumon, G. (1998). Ventilator-induced lung injury: Lessons from experimental studies. American Journal of Respiratory and Critical Care Medicine, 157, 294–323.
Webb, H. H., & Tierney, D. F. (1974). Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures: Protection by positive end-expiratory pressure. The American Review of Respiratory Disease, 110, 556–565.
Parker, J. C., Townsley, M. I., Rippe, B., Taylor, A. E., & Thigpen, J. (1984). Increased microvascular permeability in dog lungs due to high airway pressures. Journal of Applied Physiology, 57, 1809–1816.
Ricard, J. D., Dreyfuss, D., & Saumon, G. (2003). Ventilator-induced lung injury. European Respiratory Journal, 22(suppl), 2s–9s.
Plotz, F. B., Slutsky, A. S., van Vught, A. J., & Heijnen, C. J. (2004). Ventilator-induced lung injury and multiple system organ failure: A critical review of facts and hypotheses. Intensive Care Medicine, 30, 1865–1872.
Hill, J., Lindsay, T. F., Ortiz, F., Yeh, C. G., Hechtman, H. B., & Moore, F. D. (1992). Soluble complement receptor type 1 ameliorates the local and remote organ injury after intestinal ischemia-reperfusion in the rat. The Journal of Immunology, 149, 1723–1728.
Craddock, P. R., Fehr, J., Brigham, K. L., Kronenberg, R. S., & Jacob, H. S. (1980). Complement and leukocyte-mediated pulmonary dysfunction in hemodialysis. New England Journal of Medicine, 296, 769–774.
Hart, M. L., Ceonzo, K. A., Shaffer, L. A., Takahashi, K., Rother, R. P., Reenstra, W. R., et al. (2005). Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. The Journal of Immunology, 174, 6373–6380.
Bolger, M. S., Ross, D. S., Jiang, H., Frank, M. M., Ghio, A. J., Schwartz, D. A., et al. (2007). Complement levels and activity in the normal and LPS-injured lung. American Journal of Physiology-Lung Cellular and Molecular Physiology, 292, L748–L759.
Takahashi, K., Saha, D., Shattino, I., Pavlov, V. I., Stahl, G. L., Finnegan, P., et al. (2011). Complement 3 is involved with ventilator-induced lung injury. International Immunopharmacology, 11, 2138–2143.
Hammerschmidt, D. E., Weaver, L. J., Hudson, L. D., Craddock, P. R., & Jacob, H. S. (1980). Association of complement activation and elevated plasma C5a with adult respiratory distress syndrome. Pathophysiological relevance and possible prognostic value. Lancet, 1, 947–949.
Langlois, P. F., & Gawryl, M. S. (1988). Accentuated formation of the terminal C5b-9 complement complex in patient plasma precedes development of the adult respiratory distress syndrome. The American Review of Respiratory Disease, 138, 368–375.
Tsukada, H., Ying, X., Fu, C., Ishikawa, S., McKeown-Longo, P., Albelda, S., et al. (1995). Ligation of endothelial αvß3 integrin increases capillary hydraulic conductivity of rat lung. Circulation Research, 77, 651–659.
Dudek, S. M., & Garcia, J. G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. Journal of Applied Physiology, 91, 1487–1500.
Adamson, R. H., Zeng, M., Adamson, G. N., Lenz, J. F., & Curry, F. E. (2003). PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. American Journal of Physiology-Heart and Circulatory Physiology, 285, H406–H417.
Ferrara, N. (1999). Vascular endothelial growth factor: Molecular and biological aspects. Current Topics in Microbiology and Immunology, 237, 1–30.
Hocking, D. C., Phillips, P. G., Ferro, T. J., & Johnson, A. (1990). Mechanisms of pulmonary edema induced by tumor necrosis factor alpha. Circulation Research, 67, 68–77.
Carbajal, J. M., & Schaeffer, R. C., Jr. (1999). RhoA inactivation enhances endothelial barrier function. American Journal of Physiology, 277, C955–C964.
Wojciak-Stothard, B., Entwistle, A., Garg, R., & Ridley, A. J. (1998). Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell–cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. Journal of Cellular Physiology, 176, 150–165.
Hirase, T., Kawashima, S., Wong, E. Y., Ueyama, T., Rikitake, Y., Tsukita, S., et al. (2001). Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. Journal of Biological Chemistry, 276, 10423–10431.
Panetti, T. S. (2002). Differential effects of sphingosine 1-phosphate and lysophosphatidic acid on endothelial cells. Biochimica et Biophysica Acta, 1582, 190–196.
Schlegel, N., & Waschke, J. (2009). Impaired cAMP and Rac 1 signaling contribute to TNF-alpha-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation, 16, 521–533.
Soga, N., Namba, N., McAllister, S., Cornelius, L., Teitelbaum, S. L., Dowdy, S. F., et al. (2001). Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Experimental Cell Research, 269, 73–87.
Okutani, D., Han, B., Mura, M., Waddell, T. K., Keshavjee, S., & Liu, M. (2007). High-volume ventilation induces pentraxin 3 expression in multiple acute lung injury models in rats. American Journal of Physiology-Lung Cellular and Molecular Physiology, 292, L144–L153.
Liu, F., Schaphorst, K. L., Verin, A. D., Jacobs, K., Birukova, A., Day, R. M., et al. (2002). Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: Potential role of glycogen synthase kinase-3beta. The FASEB Journal, 16, 950–962.
Ricard, J. D., Dreyfuss, D., & Saumon, G. (2001). Production of inflammatory cytokines in ventilator-induced lung injury: A reappraisal. American Journal of Respiratory and Critical Care Medicine, 163, 1176–1180.
Standiford, T. J., Kunkel, S. L., Lukacs, N. W., Greenberger, M. J., Danford, J. M., Kunkel, R. G., et al. (1995). Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. The Journal of Immunology, 155, 1515–1524.
Saria, A., & Lundberg, J. M. (1983). Evans blue fluorescence: Quantitative and morphological evaluation of vascular permeability in animal tissues. Journal of Neuroscience Methods, 8, 41–49.
Green, T. P., Johnson, D. E., Marchessault, R. P., & Gatto, C. W. (1988). Transvascular flux and tissue accrual of Evans blue: Effects of endotoxin and histamine. Journal of Laboratory and Clinical Medicine, 111, 173–183.
Chen, S. F., Fei, X., & Li, S. H. (1995). A new simple method for isolation of microvascular endothelial cells avoiding both chemical and mechanical injuries. Microvascular Research, 50, 119–128.
Watanabe, H., Narai, A., & Shimizu, M. (1999). Purification and cDNA cloning of a protein derived from Flammulina velutipes that increases the permeability of the intestinal Caco-2 cell monolayer. European Journal of Biochemistry, 262, 850–857.
Dewi, B. E., Takasaki, T., & Kurane, I. (2008). Peripheral blood mononuclear cells increase the permeability of dengue virus-infected endothelial cells in association with downregulation of vascular endothelial cadherin. Journal of General Virology, 89, 642–652.
Birukova, A. A., Smurova, K., Birukov, K. G., Kaibuchi, K., Garcia, J. G., & Verin, A. D. (2006). Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvascular Research, 67, 64–77.
Csortos, C., Kolosova, I., & Verin, A. D. (2007). Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. American Journal of Physiology-Lung Cellular and Molecular Physiology, 293, L843–L854.
Tinsley, J. H., Teasdale, N. R., & Yuan, S. Y. (2004). Myosin light chain phosphorylation and pulmonary endothelial cell hyperpermeability in burns. American Journal of Physiology-Lung Cellular and Molecular Physiology, 286, L841–L847.
Yuan, Y., Huang, Q., & Wu, H. M. (1997). Myosin light chain phosphorylation: Modulation of basal and agonist-stimulated venular permeability. American Journal of Physiology, 272, H1437–H1443.
Pinhu, L., Whitehead, T., Evans, T., & Griffiths, M. (2003). Ventilator-associated lung injury. Lancet, 361, 332–340.
The Acute Respiratory Distress Syndrome Network. (2000). Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New England Journal of Medicine, 342, 1301–1308.
Ranieri, V. M., Suter, P. M., Tortorella, C., De Tullio, R., Dayer, J. M., Brienza, A., et al. (1999). Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA, 282, 54–61.
Parsons, P. E., Eisner, M. D., Thompson, B. T., Matthay, M. A., Ancukiewicz, M., Bernard, G. R., et al. (2005). Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Critical Care Medicine, 33, 1–6.
Parsons, P. E., Matthay, M. A., Ware, L. B., & Eisner, M. D. (2005). Elevated plasma levels of soluble TNF receptors are associated with morbidity and mortality in patients with acute lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology, 288, L426–L431.
Gajic, O., Dara, S. I., Mendez, J. L., Adesanya, A. O., Festic, E., Caples, S. M., et al. (2004). Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Critical Care Medicine, 32, 1817–1824.
Carroll, M. V., & Sim, R. B. (2011). Complement in health and disease. Advanced Drug Delivery Reviews, 63, 965–975.
Morgan, B. P. (2000). The complement system: An overview. Methods in Molecular Biology, 150, 1–13.
Robbins, R. A., Russ, W. D., Rasmussen, J. K., & Clayton, M. M. (1987). Activation of the complement system in the adult respiratory distress syndrome. The American Review of Respiratory Disease, 135, 651–658.
Ishikawa, S., Tsukada, H., & Bhattacharya, J. (1993). Soluble complex of complement increases hydraulic conductivity in single microvessels of rat lung. Journal of Clinical Investigation, 91, 103–109.
Preissner, K. T., Podack, E. R., & Muller-Eberhard, H. J. (1989). SC5b-7, SC5b-8 and SC5b-9 complexes of complement: Ultrastructure and localization of the S-protein (vitronectin) within the macromolecules. European Journal of Immunology, 19, 69–75.
Tedesco, F., Pausa, M., Nardon, E., et al. (1997). The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. Journal of Experimental Medicine, 185, 1619–1627.
Bossi, F., Fischetti, F., Pellis, V., et al. (2004). Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. The Journal of Immunology, 173, 6921–6927.
Kuipers, M. T., van der Poll, T., Schultz, M. J., & Wieland, C. W. (2011). Bench-to-bedside review: Damage-associated molecular patterns in the onset of ventilator-induced lung injury. Critical Care, 15, 235–245.
Bhattacharya, S., Fu, C., Bhattacharya, J., & Greenberg, S. (1995). Soluble ligands of the alpha v beta 3 integrin mediate enhanced tyrosine phosphorylation of multiple proteins in adherent bovine pulmonary artery endothelial cells. Journal of Biological Chemistry, 270, 16781–16787.
Beckers, C. M., van Hinsbergh, V. W., & van Nieuw Amerongen, G. P. (2010). Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thrombosis and Haemostasis, 103, 40–55.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (30600216/30901411) and by Second Military Medical University (05QN15), and Shanghai Education Commission (09YZ89). We would like to thank Dr. Wanyin Wang for his excellent technical assistance.
Conflict of interest
All authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
K. Liu and Y.-F. Mao have contributed equally to this work.
K. Liu and Y.-F. Mao are co-first authors.
Rights and permissions
About this article
Cite this article
Liu, K., Mao, YF., Zheng, J. et al. SC5b-9-Induced Pulmonary Microvascular Endothelial Hyperpermeability Participates in Ventilator-Induced Lung Injury. Cell Biochem Biophys 67, 1421–1431 (2013). https://doi.org/10.1007/s12013-013-9675-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-013-9675-8