
Abstract
Dengue fever has been a global health concern. Mitigation is a challenging problem due to non-availability of workable treatments. The most difficult objective is to design a perfect anti-dengue agent capable of inhibiting infections caused by all four serotypes. Various tactics have been employed in the past to discover dengue antivirals, including screening of chemical compounds against dengue virus enzymes. The objective of the current study is to investigate phytocompounds as anti-dengue remedies that target the non-structural 2B and non-structural 3 protease (NS2B-NS3pro), a possible therapeutic target for dengue fever. Initially, 300 + antiviral phytocompounds were collected from Duke’s phytochemical and ethnobotanical database and 30 phytocompounds with anti-dengue properties were identified from previously reported studies, which were virtually screened against NS2B-NS3pro using molecular docking and toxicity evaluation. The top five most screened ligands were naringin, hesperidin, gossypol, maslinic acid and rhodiolin with binding affinities of − 8.7 kcal/mol, − 8.5 kcal/mol, − 8.5 kcal/mol, − 8.5 kcal/mol and − 8.1 kcal/mol, respectively. The finest docked compounds complexed with NS2B-NS3pro were subjected for molecular dynamics (MD) simulations and binding free energy estimations through molecular mechanics generalized born surface area–based calculations. The results of the study are intriguing in the context of computer-aided screening and the binding affinities of the phytocompounds, proposing maslinic acid (MAS) as a potent bioactive antiviral for the development of phytocompound-based anti-dengue agent.
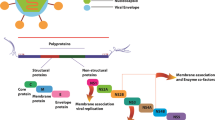
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dengue virus (DENV) spreads the dengue fever, which is the utmost transmittable mosquito-borne viral disease disturbing both the tropical and the sub-tropical region globally [1]. Dengue infection is a severe viral infection triggered by various serotypes (1, 2, 3, 4 and 5) of DENV [2] and is one amongst the seventeen “neglected tropical diseases” (NTDs) defined by the World Health Organization (WHO) [3]. The occurrence of dengue has engorged forcefully in recent years and in current days, where the endemic crossed over 100 countries, e.g. Africa, America, Malaysia, Thailand, India, Southeast Asia and Western Pacific. The initial dengue haemorrhagic fever (DHF) was reported in Thailand and Philippines in the 1950s [4] where the first two virus serotypes were recognized and tracked by the third and fourth varieties in 1954 [5]. But, the fifth serotype was isolated in October 2013 in Malaysia [6]. In India, DENV was primarily recognized in 1944 in Kolkata from the infected US soldiers by testing the serum samples. There are two well-defined appearances of DENV infection in humans, i.e. severe fever and painful symptoms (DHF/dengue shock syndrome (DSS)) [3, 7, 8]. DHF and DSS are frequently the causes of death from dengue transmission. People suffering from DHF have a 5% mortality rate, but if they switch to DSS, their mortality rate might rise to 40%. This dreadful disease has affected almost 2.5 billion people with an estimated number of 25,000 deaths per year [9]. Dengue virus belongs to the genus Flavivirus which is a Flaviviridae family member [10]. Human being and monkeys are the main hosts of DENV. It has been reported to be the existence of five different serotypes of DENV, namely DENV-1, DENV-2, DENV-3, DENV-4 and DENV-5, having antigenic differences in their genetic material. The four varieties (DENV-1, DENV-2, DENV-3 and DENV-4) are responsible for spreading the disease worldwide in a faster rate [11]. According to a report by Yung group [24], infectious serotypes were identified in 469 confirmed dengue patients, and the infection comes from 22.0% DENV-1 serotype, 57.1% DENV-2, 17.1% DENV-3 and 3.8% DENV-4. But, the infection by DENV-5 serotype was reported once in 2007 and might be having a low rate of transmission [6]. The fifth serotype was also identified in October 2013, in Sarawak, Malaysia, which initiated a new complication to control dengue [12]. This virus is vector-borne communicated to human through twin types of mosquitoes, i.e. Aedes aegypti and Aedes albopictus [13]. Even if Aedes aegypti is known as the most affecting vector, but the additional Aedes species also play a vital role in spreading the infection [3].
Since antiquity, plants with antiviral characteristics have been used to treat a wide range of ailments. A vast number of research on the antiviral efficacy of different medicinal plants against DENV have been reported in the field of antiviral drug discovery [2, 3, 8, 10, 13]. Since the past few decades, scientists have explored the natural world in search of antiviral substances that can be used against DENV. Plant-derived chemical compounds or phytocompounds have been widely employed to cure a wide range of ailments, including cancer [14, 15], HIV [16] and malaria [17]. Although DENV infection/DHF is not a novel infection, research on natural compounds including phytocompound-based treatments for it remains in its infancy. There are absolutely no specific medications existing to treat DENV infection, and the world needs a quick and robust solution to the new threat coming from the dengue infections worldwide. Therefore, a substantial study into drug discovery methodologies is required to develop effective treatments for this condition. This research aims to uncover a significant number of antiviral phytocompounds that could serve as possible therapeutic agents in order to find a cure for DENV. The current study intends to examine in silico pharmacokinetic analyses, as well as molecular docking and molecular dynamics (MD) simulations, to ensure the consistency and constancy of the phytocompounds.
DENV polyprotein structure
The genetic material of DENV is a single-stranded positive RNA, wrapped in a lipid bilayer generated by the host and encircled [18] by 180 replicas of dual glycoproteins. The single polyprotein structure is encoded by an 11-kb RNA genome. This polyprotein part is formerly sliced into three structural as well as seven non-structural proteins in the cytoplasm. The three structural proteins are involved in the protection of the genome, while the non-structural proteins are here for the biological replication process. As indicated in Fig. 1, the structural proteins are capsid (C), membrane (M) and envelope (E), and there are seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [11].

Polyprotein structure of DENV consisting of the two regions: structural and non-structural as indicated in the figure. Structural region of the polyprotein cleaves to form the capsid (C), membrane (M) and envelope (E) proteins. Non-structural part is being cleaved to form the NS1-, NS2A-, NS2B-, NS3-, NS4A-, NS4B- and NS5-like proteins. The target site of the current study is in the non-structural proteins (NS2B-NS3 region), where the NS2B part with a sequence from 43 to 104 residues and the NS3 part with residues from 18 to 167 are considered for the calculation. The target protein NS2B-NS3 is shown in a green–blue cartoon representation including the 62 residues from NS2B cofactor (green cartoon) and 150 residues for NS3 protease (blue cartoon)
Structure of non-structural 2B and non-structural 3 protease
DENV protease (non-structural 2B and non-structural 3 protease (NS2B-NS3pro)) is a trypsin-like serine protease that cleaves dengue polyprotein into the individual proteins essential for viral replication. DENV NS2B-NS3pro has been the principal target of dengue antiviral development [19]. There are two potential locations for suppressing DENV protease: the active site and the blocking attachment of protease (NS3) to its protein cofactor (NS2B). The non-structural NS3 segment is a serine protease–like trypsin, which serves a vital role in post-translation in the growth and maturation of virus [20]. This domain is made up of a conserved catalytic triad like His51, Asp75 and Ser135 [18, 21], and then the action is boosted by NS2B as the cofactor (Fig. 2). The contribution of the cofactor to the action of NS3 is mediated by its hydrophilic domain, which is responsible for retaining and promoting the initiation, while the hydrophobic area engages in association during the cleavage process [12]. Structural analysis reveals that the NS2B cofactor survives as a β-hairpin portion that envelops the NS3 protease core and contributes to the catalytically dynamic conformation of the DENV protease [18]. Dengue virus infection overthrows the immune system constructing clinical signs together with headache, inflammation, bleeding, hypertension and mental disorders where the infection terminates in death in some cases due to liver damage.

Schematic structure of DENV-2 NS2B-NS3pro with a ligand (green stick model) bound in its active site region. NS3 protease part is shown in blue cartoon, and NS2B cofactor is shown in pink carton model. Residues with importance are labelled, and hydrogen bonds (H-bonds) are shown in red-dashed lines
Reports on anti-dengue phytocompounds
Many reports of phytocompounds with anti-dengue properties have been evident from various published articles against the DENV NS2B-NS3pro. Secondary metabolites or phytocompounds are the basic source to design a drug molecule against the therapeutic target. In a detailed in silico study, Powers and Setzer [3] virtually screened a pool of phytochemicals as antiviral agents against DENV. A few alkaloids (aurones, terpenoids, lignans, chalcones, flavonoids, stilbenoids and isoflavonoids), random polyphenolic compounds (xanthones, coumarins and quinones) and incidental phytochemicals were docked with NS2B-NS3pro. Some of them conveyed fine associations towards the dynamic site of the NS2B-NS3pro like some chalcone compounds (balsacone A, balsacone B and balsacone C) [3]. In another study, phytocompounds from Endiandra kingiana were thought to be in contrast in the direction of DENV-2 NS2B-NS3 serine protease. Furthermore, molecular docking and MD simulation were used to assess the constraining strategies for the specific ligands bound to the active site’s amino acids of the serine protease [22]. In a fascinating study conducted by Trujillo-Correa et al. [23], the anti-dengue effects of 12 ethanolic extracts of designated plants obtained from an ethnobotanical assessment completed in Cartagena, Colombia, on the Colombian Caribbean coast, as well as those of 5 fractions and 5 compounds derived from Psidium guajava, were evaluated. The most selective ethanolic extracts (obtained from Psidium guajava) were fractionated, and the anti-dengue effect was confirmed by integrating the outcomes of in vitro test and in silico investigations to hypothesize prospective antivirals. In another study, a few examinations have been accounted for that, and curcumin can hinder the transmission cycles of dengue infection. In this study, a benzenesulfonyl curcumin, (3E,5E)-3,5-bis(4-methoxybenzylidene)-1-(phenylsulfonyl)piperidin-4-one, was synthesised in two steps and analysed by in silico analysis to predict the inhibitory activity of this molecule against DENV-2 NS2B-NS3pro [21].
The five virtually screened phytocompounds (gossypol (GSP), hesperidin (HSP), maslinic acid (MAS), naringin (NGN) and rhodiolin (RHO)) were investigated as DENV-2 NS2B-NS3 protease inhibitors (PIs) in the present investigation, followed by MD simulations and MM-GBSA-based free energy estimation for the specified conformation of the complexes, are being performed. The key goals of the binding energy calculation were to obtain an insight into the binding affinity of any of the chosen PIs and then to study the molecular-level interaction mechanism. It is intriguing to notice that the MAS binds to the protease with a higher affinity than the other PIs, and on account of this robust binding affinity, MAS may be well suited for the active site of the NS2B-NS3pro. Consequently, the present investigation may provide useful intuitions for designing phytocompounds as anti-dengue medicines targeting DENV NS2B-NS3pro.
Theory and methods
The current study identifies prospective anti-dengue medication candidates from specific medicinal plants previously reported in the literature, as well as antiviral phytocompounds from the Duke’s phytochemical and ethnobotanical database, which provides a graphical representation of in silico approaches implemented, are shown in supplementary Fig. S1.
Preparation of the protein
Crystal structures of NS2B-NS3pro of DENV were acquired from the Protein Data Bank (PDB). Proteases of all the four DENV serotypes with PDB IDs 3L6P, 2FOM, 3U1I and 2VBC from DENV-1, DENV-2, DENV-3 and DENV-4, respectively, were analysed in order to compare their structural similarities with each other by comparing the NS3 protease sequence information through a multiple sequence alignment study, as shown in Fig. 3A. From a 3D structure superimposition study of NS3 proteases from all four serotypes, it was confirmed that the NS3 protease’s active site region along with other important domains is mostly having similar conformation in all the four serotypes (3L6P (DENV-1), 2FOM (DENV-2), 3U1I (DENV-3) and 2VBC (DENV-4)), as depicted in Fig. 3B. The catalytic residues of active site like His51 and Ser135 remain overlapped to each other in the superimposed structure in all serotypes, suggesting some close structural conservations and similarities amongst themselves albeit sequence alignment differences. Also from the intense review of several literatures, it was found that DENV-2 is highly infectious than the other three serotypes [24, 25]. So, NS2B-NS3pro of DENV-2 was chosen, i.e. NS2BCF-Gly-Ser-Gly-NS3PRO (Fig. 1) with the PDB ID 2FOM, which is the finest structure with 1.50 Å resolution to consider as our model protein. The complete FASTA sequence of the protease from the PDB was helpful to predict the missing residues in the NS2B (cofactor) structure by using the widely used protein structure prediction server I-TASSER [26].

A Sequence alignment of NS3 protease of the four serotypes (DENV-1 (3L6P), DENV-2 (2FOM), DENV-3 (3U1I), DENV-4 (2VBC)) by ClustalW. Active site catalytic triad residues His51, Asp75 and Ser135 are highlighted in blue colour. B Superimposed cartoon structures of DENV-1 (green ribbon), DENV-2 (cyan ribbon), DENV-3 (purple ribbon) and DENV-4 (yellow ribbon) from PyMOL viewer are shown. Active site residues His51, Asp75 and Ser135 are labelled in red colour in stick model
Collection of phytocompounds through virtual screening
Collection of phytocompounds from various sources by virtual screening technique to design a proper drug candidate against dengue virus serotype 2 (NS2B-NS3pro) is the main objective of the current study. The selection process was done by picking the phytocompounds with active inhibitory nature against the NS2B-NS3pro. The process of virtual screening started from Dr. Duke’s phytochemical and ethnobotanical database. Duke’s database facilitates exhaustive searches for plants, chemicals, bioactives and ethnobotany using scientific or common names. The entire screening process of phytocompounds includes virtual screening of Duke’s database (344 antiviral phytocompounds) along with the review of literature (30 anti-dengue phytocompounds) from which five common compounds from both the sources have been considered. In the first step of virtual screening process, all those compounds were downloaded from PubChem database with their 3D structures. Their canonical SMILES were also collected from the PubChem database for toxicity and carcinogenicity test by ADMET@SAR server [27]. Amongst the whole 344 compounds, 105 compounds were selected on the basis of their human ether-a-go-go-related gene, Ames toxicity value, carcinogenicity and acute oral toxicity rate by ADMET@SAR server. In the second phase of virtual screening method, all the non-toxic and non-carcinogenic 105 compounds (as listed in supplementary Table T1) were reserved for molecular docking purpose by AutoDock tool. Four compounds, i.e. gossypol, hesperidin, naringin and maslinic acid, have been derived from the pool of 105 molecules with higher binding affinity scores towards NS2B-NS3pro active site. Again, the third segment of screening process involved a literature study of earlier investigated information from various published research papers which brought 30 phytocompounds into the knowledge with anti-dengue activity (as listed in Table 1). At this point, the interesting matter is that the four molecules, i.e. gossypol, hesperidin, naringin and maslinic acid, are present amongst those 30 molecules having strong literature evidences with anti-dengue character. Though naringin is having toxic nature, it is considered as a suitable drug molecule towards DENV NS2B-NS3pro due to its robust indication of anti-dengue behaviour [23] and also towards Zika NS2B-NS3pro [28]. Once more, naringin is reflecting developed molecular docking score and the residual interaction towards the active site of the concerned receptor, along with the non-carcinogenic stuffs. Except all these evaluations, one more molecule, i.e. rhodiolin, was found from the prior conveyed literature analysis which shows a strong inhibitory action towards dengue NS2B-NS3pro accompanied by higher docking score with non-toxicity and non-carcinogenic values by ADMET@SAR.
Modelling protease–ligand complex through docking
The crystal structure of DENV-2 NS2B-NS3pro has been retrieved from the PDB with the ID 2FOM. All the 30 phytocompounds listed in Table 1 (chosen from the earlier reports) were then screened for their anti-dengue properties through docking with the DENV NS2B-NS3pro. The PubChem database was explored for the collection of the 3D structure of the inhibitory compounds in SDF format [48, 49]. AutoDock tool (ADT) 1.5.6 [50] was then used to convert all of the selected phytocompounds to the PDBQT format by adding only polar hydrogens. Further, the desolvation of the structure was done by discarding the waters from the receptor molecule and polar hydrogens and Kollman’s charges were added for the preparation of the PDBQT format file of the macromolecules through the ADT. Tracing the model conformity of the nucleotides in the active site pocket of NS2B-NS3pro was done by applying the docking algorithm.
Grid box generation and binding site residue analysis
To determine the receptor’s active site, we used the grid box of ADT 1.5.6. The active site residues of NS2B-NS3pro studied from the literature are His51, Val52, Lys73, Asp75, Val72, Leu128, Pro132, Ser135 and Tyr150 (in the NS3 protease) and Gly151, Gly153 and Tyr161 (in the NS2B cofactor). But, the predicted catalytic triads His51, Ser135 and Asp75 exist in a pocket having catalytic effect towards the protease. The active site investigation was also carried out by using grid box analysis to calculate the x, y and z coordinates of a confined ligand with the receptor molecule. The grid centre for a NS2B-NS3pro receptor was adjusted to center_x = − 4.811, center_y = − 11.534 and center_z = 18.725 values obtained from the residue directly interacting with the protein. The dimensions of the grid’s centre were set to x = 16 Å, y = 24 Å and z = 24 Å, with a spacing of 1 Å between each grid point.
Toxicity prediction of phytocompounds
The phytocompounds were also filtered on the basis of the ADME properties by toxicity prediction by using ADMET@SAR [27]. The absorption, distribution, metabolism, excretion and toxicity (ADMET) features of entire nominated molecules are very much essential to choose a perfect drug molecule [51]. According to their toxicity effects and carcinogenic nature, the phytocompounds were chosen as drug candidates.
System setup
The input data for the assisted model building with energy refinement (AMBER) molecular mechanics curriculum has been prepared using a tleap shell script. LEaP is the fundamental tool for creating force field files such as GAFF and ff14SB. The Leap unit was applied to include all the required hydrogens in the structure. Antechamber module was taken to allocate the GAFF field constraints for the selected compounds only to process the PDB file directly generating the output files which is perfect for LEaP in AMBER 18 [Molecular mechanics generalized born surface area approach Calculation of absolute binding affinities with a modest computational exertion is done by MM/PBSA.py tool [56], which is currently used as a worldwide method and was initially developed for the AMBER software. MM/PBSA is mostly used for binding affinity studies, but nowadays, molecular mechanics generalized born surface area (MM/GBSA) is applied in most of the cases due to its faster calculation rate and comparatively higher accuracy [57]. We employed the MM-PB/GBSA method in our earlier research on HIV-1 protease [58,59,60,61] and on SARS-CoV-2 Mpro protease systems [62], and here, we reused the similar practice. In MM/GBSA, the free energy (G) calculation of a study is valued from the following equation: where Ebond = bonded (bond, angle and dihedral), Eele = electrostatic interactions, EvdW = van der Waals interactions, Gpol = polar contributions to the solvation free energy levels, Gnon-pol = non-polar contributions to the solvation free energy levels and TS = entropy effect. where ∆Gbind = the binding free energy in solution, ∆EMM = the molecular mechanics energy, ∆Gsolv = the solvation free energy and T∆S = entropy effect. The solvation free energy (∆Gsolv) is further divided into two segments. The polar solvation energy (Gpol) represents the electrostatic interaction amongst the solute and the associated solvent in the particular system. It is gained from the numerical solution of generalized-born surface area (GB). The non-polar solvation is originated from a linear relation with the solvent accessible surface area (SASA). where the MSMS software package was used with a solvent probe radius of 1.4 Å to calculate SASA. The experimental standards were established to 0.00542 kcal/mol for γ and 0.92 kcal/mol for β, individually. The solute and solvent dielectric constants remained constant at 1.0 and 80.0, respectively, in the studied system. The contribution of entropy (− T∆S) to binding affinity stands up from variations of the translational, rotational and vibrational degrees of freedom. Vibrational entropy contributions were not computed in the present study due to insufficient computational resources, the extremely time-consuming nature of the operation and the addition of rounding errors, and it is very challenging to handle the covariance fluctuation matrix of an all-atom model structure of a larger system. Due to the accurate analysis feature of generalized borne calculations in the field of computational approaches and its countless advanced criteria, GB decomposition method of MM-PBSA part in the AMBER 18 package was applied to find out the interactions between the five ligands and NS2B-NS3pro residues. Each inhibitor–residue couple comprises contribution of van der Waals interactions (∆EvdW), electrostatic interactions (∆Eele), polar solvation energy, (∆Gpol) and non-polar solvation energy (∆Gnon-pol) in the mechanism of their binding interaction effect. Total energy components in Eq. (6) were assessed, operating the 300 snapshots from the first 30-ns trajectories in all the complex systems. The polar contribution (∆Gpol) to solvation energy was calculated through the molecular mechanism of GB module and limitations for the MM/GBSA analysis developed by Amber team [63]. The constancies of the NS2B-NS3pro–ligand systems were analysed through evaluation of the H-bonds by using the hbond tool of CPPTRAJ unit [64]. A default distance cut-off of 3.0 Å and an angle cut-off of 135° have been considered, for the evaluation of bonding amongst the donor and acceptor atoms. The total numbers of H-bonds found from the entire number of frames produced by the system when get multiplied with hundred give the percentage of occupancy.Binding free energy calculation
The polar solvation term
The non-polar solvation term
Residue–ligand interface disintegration evaluation
H-bond calculation
Results and discussions
Docking of NS2B-NS3 protease with the screened ligands
A consistent, cost–benefit and quick technique is actually required in the field of in silico design of drug discovery for the docking method of the specific receptor molecules with the exploration of a large number of ligand molecules which is really possible due to the virtual screening process [65]. Finally, five phytocompounds were selected from Duke’s database and previously published reports as listed in Table 1 with anti-dengue properties against NS2B-NS3pro.
Based on ADMET test, binding free energy results and interaction with the important active site residues, we finally selected GSP [3], HSP [23], MAS [32], NGN [23] and RHO [3]. These compounds come along with Lipinski’s rule of five and have a well interaction with the active site residues of the NS2B-NS3pro. During the screening of phytocompounds, these compounds were found to be in Duke’s database having antiviral nature. The five compounds with PubChem CID (3503 (gossypol), 442,428 (naringin), 10,621 (hesperidin), 73,659 (maslinic acid) and 14,778,358 (rhodiolin), the 2D structures of which are shown in Fig. 4, were selected based on their higher affinity score and active site residue-interacting feature with the protease (Fig. 5). The five phytocompounds (GSP, NGN, HSP, MAS and RHO) acting as inhibitors against NS2B-NS3pro have been docked through ADT, carrying the binding affinity of − 8.5, − 8.7, − 8.5, − 8.5 and − 8.1 kcal/mol, respectively, as shown in Table 2.

Two-dimensional molecular structures of the five screened phytocompounds (A GSP, B HSP, C MAS, D NGN and E RHO), which are studied against the DENV NS2B-NS3pro

The details of interacting residues present near to the active site of NS2B-NS3pro with the five drug candidates (GSP, HSP, MAS, NGN and RHO). Ligands/phytocompounds are shown in grey stick model, and residues of interactions are displayed in dyed spheres. Green spheres: conventional H-bonds; cyan spheres: C–H-bonds; red spheres: unfavourable donor–donor, unfavourable acceptor–acceptor; light pink spheres: alkyl and pi-alkyl; purple sphere: pi-sigma; pink sphere: pi-pi stacked
Surface view of the protease–ligand complexes is shown in Fig. 6, which shows the interaction of the NS2B-NS3pro with all the five inhibitor compounds in its active site region. Taking this figure as our concern, we found a number of interacting residues in the active site pocket, viz. His51, Arg54 and Ser135 (Fig. 6A, NS2B-NS3pro/GSP complex); Val72, Lys73, Asp75, Ser135, Asn152 and Gly153 (Fig. 6B, NS2B-NS3pro/HSP complex); His51 (Fig. 6C, NS2B-NS3pro/MAS complex); His51, Pro132, Ser135 and Asn152 (Fig. 6D, NS2B-NS3pro/NGN complex); and His51, Asp75, Pro132, Ser135 and Gly153 (Fig. 6E, NS2B-NS3pro/RHO complex).

Predictable binding approaches found from the docking study of all the five inhibitors with the DENV protease (NS2B-NS3.pro) displayed as a molecular surface model in grey, and the colourful sticks are the inhibitor molecules, i.e. A GSP (green), B HSP (deep blue), C MAS (yellow), D NGN (cyan) and E RHO (pink)
Toxicity evaluation
Numerous drug improvement matters are regarded as serious aspects that will have a key stimulus upon computer-based drug designing field in medicinal chemistry’s current progression in the present era. Evaluation of the toxicity rate of the five phytocompounds through the ADMET@SAR was done with five different pharmacokinetic properties like promising absorption, leading distribution, governing metabolism, supporting excretion and evading toxicity. Predictions of the classification and regression values were completed for the lead molecules by this server. The calculated results were taken on various bases like human ether-a-go-go-related gene, inhibition, Ames toxicity, carcinogens, biodegradation, acute oral toxicity and carcinogenicity as shown in Table 3. According to the acute oral toxicity value of the selected phytocompounds, categories III and IV can be considered as a drug candidate. Though naringin is an Ames toxic compound, its binding affinity is − 8.7 kcal/mol which is highest amongst the five selected molecules along with the acute oral toxicity level III. Taking the active site interaction as our main concern (check Table 2), we included naringin as an inhibitory molecule in our study.
RMSD: conformational stability analysis
The protease NS2B-NS3 complexed with the five drug candidates was subjected for MD simulations for 50 ns in order to study the variation in the conformational stability of the protein as well as its dynamic nature. Estimation of the RMSD of the Cα atoms has been already done and is presented in Fig. 7, The RMSD plot displays that the conformations of the NS2B-NS3pro complexed with GSP, HSP, MAS and NGN are in a stable state, maintaining a decent steadiness along with a good equilibrium owing to their low RMSDs. These four trajectories run equivalent to each other till the completion of the simulations, fluctuating from 2.0 Å to a highest of 3.5 Å. The path for the NS2B-NS3pro/RHO complex varies a bit more after completion of 1-ns simulation jum** to as high as 4.5 Å, though in due course, it comes downcast to go parallel with others, suggesting stable simulations. The average RMSD values were 2.51 Å, 2.84 Å, 2.62 Å, 3.83 Å and 2.78 Å for the complexes GSP, HSP, MAS, NGN and RHO, respectively, signifying an advanced rate of conformational stability in each of the protease–ligand complex.

A plotting of root-mean-square deviation for backbone Cα atoms comparative to the preliminary minimized complex system (NS2B-NS3 protease + phytocompounds) as a function of time
Rg: the conformational compactness analysis
The conformational stability of the trajectories was studied through esteeming the radius of gyration (Rg) from their consistent simulation routes, and the projected average values are labelled in supplementary Fig. S2. Rg is a parameter to show the initial compactness of the protein structure and its changes in compactness during the simulation phase. A comparable Rg is accomplished for every system, suggesting a similar flexibility and compactness for all the complexes.
Root-mean-square fluctuation: the conformational flexibility analysis
To analyse the detail fluctuations of the residual atoms, the root-mean-square fluctuation (RMSF) of the Cα atoms of whole residues was estimated and is revealed in Fig. 8.

The root-mean-square fluctuations of Cα atoms vs. the residue number of the phytocompound composite NS2B-NS3pro assembly. Residues having higher fluctuations and larger than 3.0 Å are labelled by a cut-off black-dashed line and are labelled. Two regions (Ile78-Met84 and Gly98-Gly104) of the NS2B cofactor circled by blue-dashed lines show the highest flexibility for the protein
RMSF values of the NS2B-NS3pro structure confirmed that the NS2B cofactor residues exhibited higher fluctuations, suggesting increased dynamics in the region. The average RMSF values were 1.41 Å, 1.52 Å, 1.51 Å, 1.43 Å and 1.50 Å for the complexes GSP, HSP, MAS, NGN and RHO, respectively, signifying an advanced rate of conformational variations in the protease complex, whereas the average RMSF values for the NS2B cofactor region only were 1.96 Å, 2.50 Å, 2.38 Å, 2.90 Å and 2.14 Å in the complexes with GSP, HSP, MAS, NGN and RHO, respectively, signifying a higher rate of conformational fluctuations in the NS2B cofactor region with NGN-complexed structures and lower rate with the GSP-complexed structures. From Fig. 8, it was observed that the residues (Gly43-Glu48, Ile78-Met84 and Gly98-Gly104) in the NS2B cofactor have higher fluctuations as compared to those in the NS3 protease, of which residues have as high as a RMSF of 3.03 Å (Thr120 in the NS2B-NS3pro/NGN complex).
Binding free energy analysis
As shown in Fig. 9 and Table 4, the calculated binding free energy levels (ΔGtotal) of five compounds (GSP, HSP, MAS, NGN and RHO) were − 22.48 kcal/mol, − 13.30 kcal/mol, − 29.20 kcal/mol, − 16.45 kcal/mol and − 15.76 kcal/mol, respectively. This implies that binding free energy levels of all the five complexes studied are either reasonably higher than or approximately equal with the binding affinities of the other NS2B-NS3pro inhibitors reported earlier like ganodermanontriol (− 24.46 kcal/mol), lucidumol (− 19.73 kcal/mol), ganoderic acid C2 (− 19.04 kcal/mol), ganosporeric acid (− 11.45 kcal/mol) [50] and panduratin A (− 11.27 kcal/mol), and thioguanine-based inhibitors like compound 18 (− 16.37 kcal/mol), compound 21 (− 20.95 kcal/mol) [12], cyanidin 3-glucoside (− 9.04 kcal/mol), dithymoquinone (− 11.74 kcal/mol) and glabridin (− 28.71 kcal/mol) [51].According to the component analysis of the binding free energy from Table 4, in all the five NS2B-NS3pro/inhibitor systems, van der Waals (EvdW) and electrostatic energy levels (Eele) in the gas phase offer the significant contributions to the inhibitor binding. Non-polar solvation energy levels (ΔGnon-pol) arose from the SASA of the interment of ligand’s zone, which has a deep impact to the binding energy in a favourable mode. On the contrary, polar solvation energy levels (ΔGpol) produce a substantial unfavourable contribution to the binding energy. In case of all the four complexes (except NS2B-NS3pro/HSP complex), the van der Waals interaction (ΔEvdW) is more reasonable as compared to the intermolecular electrostatic interactions (ΔEele). Furthermore, Table 4 represents the estimated binding free energy (ΔGtotal) of MAS (− 29.20 kcal/mol) is superior than that of GSP (− 22.48 kcal/mol), NGN (− 16.45 kcal/mol), RHO (− 15.76 kcal/mol) and HSP (− 13.30 kcal/mol), signifying that MAS is more active against NS2B-NS3pro than the other four considered phytocompounds.

Energy components (kcal/mol) for the binding of inhibitors to the NS2B-NS3pro. ΔEvdW, van der Waals energy; ΔEele, electrostatics energy in the gas phase; ΔGpol, polar solvation energy; ΔGnon-pol, non-polar solvation energy; and ΔGtotal, total binding energy
From the review of earlier reports [12, 50, 51], it was found that the estimated binding free energy for glabridin (− 28.71 kcal/mol) and ganodermanontriol (− 24.465 kcal/mol) is higher as compared to that for the other phytocompounds listed in Table 5. However, in our study, MAS was found to be the phytocompound with the highest binding affinity for NS2B-NS3 protease with − 29.20 kcal/mol, which can be compared to the compounds like glabridin and ganodermanontriol. Conversely, it was observed that cyanidin 3-glucoside (− 9.04 kcal/mol) and panduratin A (− 11.27 kcal/mol) have the lowest binding affinities amongst all the listed compounds (Table 5). Moreover, in the current study, HSP has the lowest binding affinity, i.e. − 13.30 kcal/mol, amongst the five inhibitor phytocompounds and with higher binding affinity compared to certain compounds like ganosporeric acid, panduratin A and cyanidin 3-glucoside. The data comparison from Table 4 and Table 5 provides a strong evidence about the potential binding affinity of our screened phytocompounds which falls in the resulting order against NS2B-NS3pro as follows: maslinic acid > glabridin > ganodermanontriol > gossypol > compound 21 > lucidumol > ganoderic acid C2 > naringin > compound 18 > rhodiolin > hesperidin > dithymoquinone > ganosporeric acid > panduratin A > cyanidin 3-glucoside. Hence, the phytocompounds like MAS (− 29.20 kcal/mol) and GSP (− 22.48 kcal/mol) may be reflected as a principal component in the finding of rational drugs against DENV NS2B-NS3pro.
Structure–affinity relationship analysis
The structure–activity relationship was investigated utilising the independent residue decomposition of free energy using MM-GBSA to identify and determine the hotspot amino acid residues involved in the complexes’ binding procedure.
As shown in Fig. 10, the binding free energy can be decomposed into inhibitor–residue sets to form a well interaction scale. Residues with interaction energy more than –1.0 kcal/mol are considered to be the hotspot residues and believed to be taking part in the binding process of the compounds. The residue decomposition study was used to better understand drug resistance methods at the molecular level, as well as to determine the involvement of particular residues in protein–inhibitor interactions. Figure 10 proves that complete frameworks in the interaction bands of five NS2B-NS3pro/phytocompound complexes stay related, and still there is a difference in distinct residue interaction spectrum. Globally, the main interactions come from a cluster of hotspot residues like His51, Leu128, Pro132 and Tyr161, which contributes favourably to the binding incident. Residues like Ile36, Val52, Arg54, Gly133, Ser135 and Val154 also collectively contribute for the binding. Nevertheless, the unfavourable polar solvation energy levels in aqueous solution contradict the net binding energy levels.

Decomposition of ΔG on a per-residue basis for the NS2B-NS3.pro–ligand complex (GSP, HSP, MAS, NGN and RHO)
Figure 10 and Table 6 reveal that the complete frameworks in the interaction peaks of five NS2B-NS3pro–ligand structures are comparable, despite differences in specific nucleotide interaction peaks. On the whole, the crucial interactions are caused by a cluster of hotspot nucleotides such as Ile36, His51, Leu128, Pro132, Val154, Val155, Tyr161 and Lys173, all of which positively contribute to the binding event. Furthermore, the residues in the catalytic site (His51, Leu128 and Pro132) contribute greatly (> 1.0 kcal) to the binding of all inhibitors (except HSP) to the protease.
The decomposition of ΔG values on a per-residue basis is depicted in Fig. 11. ΔG values are split per residue into contributions from van der Waals (EvdW), the sum of electrostatic interactions in the gas phase and polar solvation energy (ΔGpol = ΔEele + ΔGgb) and non-polar solvation energy (ΔGnon-pol) for residues with ΔG ≥ 1.0 kcal/mol for all the five complexes. Furthermore, it can be noted that the catalytic residues Tyr161 and Pro132 contribute significantly (≥ 1.0 kcal) to the binding of all four inhibitors to the protease, excluding hesperidin. The unfavourable polar solvation energy levels cancel out the net binding energy levels due to their aqueous solution solvation. Residue like Lys173 has relatively unfavourable polar solvation energy levels with ≥ 1 kcal/mol. Table 6 further divides per-residue contributions into backbone atoms and side chain atoms.

Decomposition of ΔG on a per-residue basis into contributions from the van der Waals energy (EvdW), the sum of electrostatic interactions and polar solvation energy (ΔEele + ΔGpol) and non-polar solvation energy (ΔGnon-pol) for the residues of |ΔG|≥ 1.0 kcal/mol. NS2B-NS3pro–gossypol, NS2B-NS3pro–hesperidin, NS2B-NS3pro–naringin, NS2B-NS3pro–rhodiolin and NS2B-NS3pro–maslinic acid complexes from top to bottom
Two-dimensional structure visualization through LigPlot +
A wide-ranging interaction summary of residues regarding H-bonds and hydrophobic interactions was also estimated through the LigPlot + software package [68] and is presented in Fig. 12. It specifies that many of the residues present exactly in the active site pocket and nearby this region play a notable action in evolving H-bonds by means of the inhibitory compounds. Analysis of the hydrophobic and H-bond interactions of the whole residues of the individual system was calculated by LigPlot + software. Here, the residues like Asp75, Phe130 and Tyr150 (GSP complex); Phe130, Tyr150, Gly151 and Gly153 (HSP complex); Asp75 (MAS complex); and Gly151 (RHO complex) are present nearby the active site pocket having H-bonds with the inhibitors and also have a major role in this interaction. Apart from the H-bonds, many residues in the active site or its surrounding region contribute through hydrophobic interactions as in case of NGN complex (Fig. 12).

MD simulation analyses of all five phytocompounds with the NS2B-NS3 protease revealed the expected binding modes. The phytocompounds are denoted as stick, and their interacting residues in the protease are designed by the LigPlot + software package. The bonds formed between phytocompounds and NS2B-NS3 pro residues are labelled in red. A NS2B-NS3pro–GSP complex. B NS2B-NS3pro–HSP complex. C NS2B-NS3pro–MAS complex. D NS2B-NS3pro–NGN. E NS2B-NS3pro–RHO complex
H-bond analysis
To supplement the phytocompounds’ binding affinities, the binding stabilities in all five complexes with the NS2B-NS3pro structure were examined during the 50-ns MD simulation trajectory, and the H-bond occupancy is explained in supplementary Table T2. Figure 13 represents the number of H-bonds as a function of time, and it is discovered that the protein–ligand complex of HSP with the NS2B-NS3pro structure has the highest average number of H-bonds (23.9) per time frame during the simulation phase. The average numbers of H-bonds witnessed for GSP, MAS, NGN and RHO were 21.5, 20.7, 23.0 and 21.1, respectively. The study suggests that for all the phytocompounds, at least 20 hydrogen bonds are always present throughout the simulation period, indicating that the inhibitors have close interactions with the protease active site region. Data analysis of Table T2 reveals that residues like PHE_130@O (48.7%, 29.5%) and ASP_75@OD2, TYR_150@OH and ASP_75@OD2 (25.4%, 23.4% and 14.7%, respectively) donate in the complex NS2B-NS3pro/GSP to make hydrogen bonds with the ligand with a possession of minimum 10% all over the phase of MD simulation. On the contrary, for example in the NS2B-NS3pro/HSP composite, the extreme occupancy by hydrogen bonding is attained by the residues ASP_75@OD1 and ASP_75@OD2 (55.2%, 55.1%, 39.7% and 38.6%). Moreover, in the case of NS2B-NS3pro/MAS, there were at least two residues: HIS_51@O contributes 31.0%, ARG_54@NH2 provides 16.5% and ASP_75@OD1 contributes 12.4% significantly for the bonding. Overall, all the catalytic triad residues (His51, Asp75 and Ser135) participate in the process of H bonding, making the interactions of MAS with the NS2B-NS3pro stronger. Furthermore, in the case of NS2B-NS3Pro/NGN, residues like ILE_36@O, GLY_153@O, PRO_132@O and PHE_130@O donate 37.4%, 34.0%, 15.3% and 11.2%, respectively. Lastly, in the case of NS2B-NS3pro/RHO, residues namely ASP_75@OD1 and ILE_36@O, ASP_75@OD2 and GLN_27@O donate significantly thru 33.6% and 25.8%, 22.2% and 20.1%, respectively. From these analysis (Table T2 and Fig. 13), we found the H bonding patterns in instance of the protease/ligand complexes accomplished through establishment of at least two H-bonds (for GSP, HSP, MAS, NGN and RHO) exist throughout the simulation period with well and average pattern.

Average intermolecular hydrogen bonding numbers and variations in NS2B-NS3pro with inhibitors gossypol, hesperidin, naringin, rhodiolin and maslinic acid, complexes during 50-ns MD simulations. The y-axis shows the number of H-bonds, and the x-axis shows the simulation time in ns
Conclusion
The present work supports the use of phytocompounds as an anti-dengue agent targeting the NS2B-NS3pro of DENV. Here, we report for the first time the efficacy of inhibitory properties of the phytocompounds MAS, GSP, NGN, RHO and HSP. Moreover, our research work revealed that most of these phytocompounds are effective enough as compared to the previously reported synthetic NS2B-NS3pro inhibitors or phytochemicals owing to their binding efficacy and sustaining capacity to persist in the active site for up to a nanosecond time scale. The complete investigation of all the five selected inhibitors, viz. gossypol, hesperidin, maslinic acid, naringin and rhodiolin, and its mechanism of binding to the active site region of NS2B-NS3pro has done by applying MD simulations of 50 ns along with MM/GBSA analysis. In contrast with the energy components of the binding affinity, designed for the entire five NS2B-NS3pro + phytocompound complexes, the van der Waals (EvdW) energy levels and electrostatic (Eele) energy levels contribute the prime activity to the phytocompound binding, along with the non-polar solvation free energy. MAS is comparatively more effective than GSP, HSP, NGN and RHO for an enhanced favourable participation from the intermolecular van der Waals and decreased polar solvation according to our predictions. Further, MAS interacts with the catalytic residue His51 constantly throughout the simulation period, indicating its persistent residency in the active site. Our conclusion shows the direction of the priorities of evolving medications against the DENV with these experimental phytocompounds especially like maslinic acid, which is really convincing. An enhanced experimental analysis on this way of drug designing is needed by the exploration and estimation of these phytocompounds against NS2B-NS3pro in a vast range.
Data availability
All the data are available in the manuscript and supplementary information.
Code availability
Not applicable.
Abbreviations
- ADMET:
-
Absorption, distribution, metabolism, excretion and toxicity
- ADT:
-
AutoDock tool
- DENV:
-
Dengue virus
- DHF:
-
Dengue haemorrhagic fever
- DSS:
-
Dengue shock syndrome
- GSP:
-
Gossypol
- HSP:
-
Hesperidin
- MAS:
-
Maslinic acid
- MM/GBSA:
-
Molecular mechanics generalized born surface area
- MM/PBSA:
-
Molecular mechanics Poisson–Boltzmann surface area
- NGN:
-
Naringin
- NS2B-NS3pro :
-
Non-structural 2B and non-structural 3 protease
- NTDs:
-
Neglected tropical diseases
- PDB:
-
Protein Data Bank
- PME:
-
Particle mesh Ewald
- R g :
-
Radius of gyration
- RHO:
-
Rhodiolin
- RMSD:
-
Root-mean-square deviation
- RMSF:
-
Root-mean-square fluctuation
References
Mirza SB, Salmas RE, Fatmi MQ, Durdagi S (2016) Virtual screening of eighteen million compounds against dengue virus: combined molecular docking and molecular dynamics simulations study. J Mol Graph Model 66:99–107. https://doi.org/10.1016/j.jmgm.2016.03.008
Rasool N, Ashraf A, Waseem M, Hussain W, Mahmood S (2019) Computational exploration of antiviral activity of phytochemicals against NS2B/NS3 proteases from dengue virus. Turkish Journal of Biochemistry 44:261–277. https://doi.org/10.1515/tjb-2018-0002
Powers CN, Setzer WN (2016) An in-silico investigation of phytochemicals as antiviral agents against dengue fever. Comb Chem High Throughput Screen 19:516–536. https://doi.org/10.2174/1386207319666160506123715
Guha-Sapir D, Schimmer B (2005) Dengue fever: new paradigms for a changing epidemiology. Emerg Themes Epidemiol 2:1. https://doi.org/10.1186/1742-7622-2-1
Kanna SU, Krishnakumar N (2019) Anti-dengue medicinal plants: a mini review. Journal of Pharmacognosy and Phytochemistry 8:4245–4249
Mustafa MS, Rasotgi V, Jain S, Gupta V (2015) Discovery of fifth serotype of dengue virus (DENV-5): a new public health dilemma in dengue control. Med J Armed Forces India 71:67–70. https://doi.org/10.1016/j.mjafi.2014.09.011
Loroño-Pino MA, Cropp CB, Farfán JA, Vorndam AV, Rodríguez-Angulo EM, Rosado-Paredes EP et al (1999) Common occurrence of concurrent infections by multiple dengue virus serotypes. Am J Trop Med Hyg 61:725–730. https://doi.org/10.4269/ajtmh.1999.61.725
Malabadi R, Chalannavar R, Supriya S, Nityasree B, Sowmyashree K, Meti N (2018) Role of botanical drugs in controlling dengue virus disease. International Journal of Research and Scientific Innovations 5:134–159
Stanaway JD, Shepard DS, Undurraga EA, Halasa YA, Coffeng LE, Brady OJ et al (2016) The global burden of dengue: an analysis from the Global Burden of Disease Study 2013. Lancet Infect Dis 16:712–723. https://doi.org/10.1016/s1473-3099(16)00026-8
Panraksa P, Ramphan S, Khongwichit S, Smith DR (2017) Activity of andrographolide against dengue virus. Antiviral Res 139:69–78. https://doi.org/10.1016/j.antiviral.2016.12.014
Meng F, Badierah RA, Almehdar HA, Redwan EM, Kurgan L, Uversky VN (2015) Unstructural biology of the dengue virus proteins. Febs j 282:3368–3394. https://doi.org/10.1111/febs.13349
Hariono M, Choi SB, Roslim RF, Nawi MS, Tan ML, Kamarulzaman EE et al (2019) Thioguanine-based DENV-2 NS2B/NS3 protease inhibitors: virtual screening, synthesis, biological evaluation and molecular modelling. PLoS ONE 14:e0210869. https://doi.org/10.1371/journal.pone.0210869
Perera SD, Jayawardena UA, Jayasinghe CD (2018) Potential use of Euphorbia hirta for dengue: a systematic review of scientific evidence. J Trop Med 2018:2048530. https://doi.org/10.1155/2018/2048530
Sophia J, Kiran Kishore TK, Kowshik J, Mishra R, Nagini S (2016) Nimbolide, a neem limonoid inhibits phosphatidyl inositol-3 kinase to activate glycogen synthase kinase-3beta in a hamster model of oral oncogenesis. Sci Rep 6:22192. https://doi.org/10.1038/srep22192
Hsueh KC, Lin CL, Tung JN, Yang SF, Hsieh YH (2018) Nimbolide induced apoptosis by activating ERK-mediated inhibition of c-IAP1 expression in human hepatocellular carcinoma cells. Environ Toxicol 33:913–922. https://doi.org/10.1002/tox.22576
Panda M, Priyanka P, Meher BR (2022) Structure-based virtual screening, ADMET profiling, and molecular dynamics simulation studies on HIV-1 protease for identification of active phytocompounds as potential anti-HIV agents. Mol Simul 48:19. https://doi.org/10.1080/08927022.2022.2060968
Satish PVV, Kumari DS, Sunita K (2017) Antiplasmodial efficacy of Calotropisgigantea (L.) against Plasmodium falciparum (3D7 strain) and Plasmodium berghei (ANKA). J Vector Borne Dis 54:215–25. https://doi.org/10.4103/0972-9062.217612
Noble CG, Seh CC, Chao AT, Shi PY (2012) Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol 86:438–446. https://doi.org/10.1128/jvi.06225-11
Lim SYM, Chieng JY, Pan Y (2021) Recent insights on anti-dengue virus (DENV) medicinal plants: review on in vitro, in vivo and in silico discoveries. All Life 14:1–33
Kronenberger T, SMS M, Kumar Tonduru A, GonçalvesMaltarollo V, Poso A (2021) Ligand accessibility insights to the dengue virus NS3-NS2B protease assessed by long-timescale molecular dynamics simulations. ChemMedChem 16:2524–34. https://doi.org/10.1002/cmdc.202100246
Zamri A (2019) Synthesis and in silico studies of a benzenesulfonyl curcumin analogue as a new anti dengue virus type 2 (DEN2) NS2B/NS3. Indonesian Journal of Pharmacy 30:84–90
Sulaiman SN, Hariono M, Salleh HM, Chong S-L, Yee LS, Zahari A et al (2019) Chemical constituents from Endiandra kingiana (Lauraceae) as potential inhibitors for dengue type 2 NS2B/NS3 serine protease and its molecular docking. Nat Prod Commun 14:1934578X19861014
Trujillo-Correa AI, Quintero-Gil DC, Diaz-Castillo F, Quiñones W, Robledo SM, Martinez-Gutierrez M (2019) In vitro and in silico anti-dengue activity of compounds obtained from Psidium guajava through bioprospecting. BMC Complement Altern Med 19:298. https://doi.org/10.1186/s12906-019-2695-1
Yung CF, Lee KS, Thein TL, Tan LK, Gan VC, Wong JGX et al (2015) Dengue serotype-specific differences in clinical manifestation, laboratory parameters and risk of severe disease in adults. Singapore Am J Trop Med Hyg 92:999–1005. https://doi.org/10.4269/ajtmh.14-0628
Galula JU, Shen WF, Chuang ST, Chang GJ, Chao DY (2014) Virus-like particle secretion and genotype-dependent immunogenicity of dengue virus serotype 2 DNA vaccine. J Virol 88:10813–10830. https://doi.org/10.1128/JVI.00810-14
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. https://doi.org/10.1038/nprot.2010.5
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G et al (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105. https://doi.org/10.1021/ci300367a
Lim HJ, Nguyen TT, Kim NM, Park JS, Jang TS, Kim D (2017) Inhibitory effect of flavonoids against NS2B-NS3 protease of ZIKA virus and their structure activity relationship. Biotechnol Lett 39:415–421. https://doi.org/10.1007/s10529-016-2261-6
Chiow KH, Phoon MC, Putti T, Tan BK, Chow VT (2016) Evaluation of antiviral activities of Houttuynia cordata Thunb. extract, quercetin, quercetrin and cinanserin on murine coronavirus and dengue virus infection. Asian Pac J Trop Med. 9:1–7. https://doi.org/10.1016/j.apjtm.2015.12.002
Patel NK, Jaiswal G, Bhutani KK (2016) A review on biological sources, chemistry and pharmacological activities of pinostrobin. Nat Prod Res 30:2017–2027. https://doi.org/10.1080/14786419.2015.1107556
Modak D, Guha SK (2012) Symmetrical peripheral gangrene: a rare complication of dengue fever. Indian J Med Sci 66:292–295
Suganya G, Karthi S, Shivakumar MS (2014) Larvicidal potential of silver nanoparticles synthesized from Leucas aspera leaf extracts against dengue vector Aedes aegypti. Parasitol Res 113:875–880. https://doi.org/10.1007/s00436-013-3718-3
Kaushik S, Dar L, Kaushik S, Yadav JP (2021) Anti-dengue activity of super critical extract and isolated oleanolic acid of Leucas cephalotes using in vitro and in silico approach. BMC Complement Med Ther 21:227. https://doi.org/10.1186/s12906-021-03402-2
Gao Y, Tai W, Wang N, Li X, Jiang S, Debnath AK et al (2019) Identification of novel natural products as effective and broad-spectrum anti-Zika virus inhibitors. Viruses. 11. https://doi.org/10.3390/v11111019
Hussain W, Qaddir I, Mahmood S, Rasool N (2018) In silico targeting of non-structural 4B protein from dengue virus 4 with spiropyrazolopyridone: study of molecular dynamics simulation. ADMET and virtual screening Virusdisease 29:147–156. https://doi.org/10.1007/s13337-018-0446-4
Hidari KI, Takahashi N, Arihara M, Nagaoka M, Morita K, Suzuki T (2008) Structure and anti-dengue virus activity of sulfated polysaccharide from a marine alga. Biochem Biophys Res Commun 376:91–95. https://doi.org/10.1016/j.bbrc.2008.08.100
Keivan Z, Teoh B-T, Sam S-S, Wong P-F, Mustafa MR, AbuBakar S (2011) In vitro antiviral activity of fisetin, rutin and naringenin against dengue virus type-2. Journal of Medicinal Plants Research 5:5534–5539
Zandi K, Teoh BT, Sam SS, Wong PF, Mustafa MR, Abubakar S (2011) Antiviral activity of four types of bioflavonoid against dengue virus type-2. Virol J 8:560. https://doi.org/10.1186/1743-422x-8-560
Elkhalifa D, Al-Hashimi I, Al Moustafa AE, Khalil A (2021) A comprehensive review on the antiviral activities of chalcones. J Drug Target 29:403–419. https://doi.org/10.1080/1061186x.2020.1853759
Sivakumar D, Richa T, Rajesh SS, Gorai B, Sivaraman T (2012) In silico methods for designing antagonists to anti-apoptotic members of Bcl-2 family proteins. Mini Rev Med Chem 12:1144–1153. https://doi.org/10.2174/138955712802762202
Paemanee A, Hitakarun A, Wintachai P, Roytrakul S, Smith DR (2019) A proteomic analysis of the anti-dengue virus activity of andrographolide. Biomed Pharmacother 109:322–332. https://doi.org/10.1016/j.biopha.2018.10.054
Li F, Khanom W, Sun X, Paemanee A, Roytrakul S, Wang D et al (2020) Andrographolide and its 14-aryloxy analogues inhibit Zika and dengue virus infection. Molecules. 25. https://doi.org/10.3390/molecules25215037
**ao X, Si X, Tong X, Li G (2011) Preparation of flavonoids and diarylheptanoid from Alpinia katsumadai hayata by microwave-assisted extraction and high-speed counter-current chromatography. Sep Purif Technol 81:265–269
Kutumbarao N, Ramakrishnan C, Balasubramanian K, Velmurugan D (2016) Computational assessment of inhibitory activity of acridone, xanthone and flavone derivatives against NS2B/NS3pro of dengue virus type 2. J Emerg Dis Virol 2:2473–1846
Ribeiro KA, de Carvalho CM, Molina MT, Lima EP, López-Montero E, Reys JR et al (2009) Activities of naphthoquinones against Aedes aegypti (Linnaeus, 1762) (Diptera: Culicidae), vector of dengue and Biomphalaria glabrata (Say, 1818), intermediate host of Schistosoma mansoni. Acta Trop 111:44–50. https://doi.org/10.1016/j.actatropica.2009.02.008
Brandão CM, Cavalcante KSB, de Teles RM, de Marques GEC, Monteiro OS, Andrade EHA et al (2020) Composition and larvicidal activity of the oil of Dizygostemon riparius (Plantaginaceae), a new aromatic species occurring in Maranhão, Brazil. Chem Biodivers 17:e2000462. https://doi.org/10.1002/cbdv.202000462
Gade S, Rajamanikyam M, Vadlapudi V, Nukala KM, Aluvala R, Giddigari C et al (2017) Acetylcholinesterase inhibitory activity of stigmasterol & hexacosanol is responsible for larvicidal and repellent properties of Chromolaena odorata. Biochim Biophys Acta Gen Subj 1861:541–550. https://doi.org/10.1016/j.bbagen.2016.11.044
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S et al (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 47:D1102–D1109. https://doi.org/10.1093/nar/gky1033
Sharma P, Joshi T, Joshi T, Chandra S, Tamta S (2021) Molecular dynamics simulation for screening phytochemicals as α-amylase inhibitors from medicinal plants. J Biomol Struct Dyn 39:6524–6538. https://doi.org/10.1080/07391102.2020.1801507
Goodsell DS, AO AJ (1990) Automated docking of substrates to proteins by simulated annealing. Proteins: Structure, Function and Genetics. 8.
Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z et al (2019) admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 35:1067–9. https://doi.org/10.1093/bioinformatics/bty707
Case, D.A., I.Y. Ben-Shalom, S.R. Brozell, D.S. Cerutti, T.E. Cheatham, III, V.W.D. Cruzeiro, T.A. Darden,, R.E. Duke, D.G., M.K. Gilson, H. Gohlke, A.W. Goetz, D. Greene, R Harris, N. Homeyer, Y. Huang,, S. Izadi, A.K., T. Kurtzman, T.S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D.J., Mermelstein, K.M.M., Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R., Qi, D.R.R., A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C.L. Simmerling, J. Smith, R. Salomon-, Ferrer, J.S., R.C. Walker, J. Wang, H. Wei, R.M. Wolf, X. Wu, L. **ao, D.M. York and P.A. Kollman (2018) AMBER 2018, University of California, San Francisco
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A (1984) Haak JR Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 81:3684–90. https://doi.org/10.1063/1.448118
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. https://doi.org/10.1016/0021-9991(77)90098-5
Miller BR 3rd, McGee TD Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 8:3314–21. https://doi.org/10.1021/ct300418h
Krieger E, Nielsen JE, Spronk CA, Vriend G (2006) Fast empirical pKa prediction by Ewald summation. J Mol Graph Model 25:481–486. https://doi.org/10.1016/j.jmgm.2006.02.009
Meher BR, Wang Y (2012) Interaction of I50V mutant and I50L/A71V double mutant HIV-protease with inhibitor TMC114 (darunavir): molecular dynamics simulation and binding free energy studies. J Phys Chem B 116:1884–1900. https://doi.org/10.1021/jp2074804
Meher BR, Wang Y (2012) Binding of single walled carbon nanotube to WT and mutant HIV-1 proteases: analysis of flap dynamics and binding mechanism. J Mol Graph Model 38:430–445. https://doi.org/10.1016/j.jmgm.2012.10.001
Meher BR, Wang Y (2015) Exploring the drug resistance of V32I and M46L mutant HIV-1 protease to inhibitor TMC114: flap dynamics and binding mechanism. J Mol Graph Model 56:60–73. https://doi.org/10.1016/j.jmgm.2014.11.003
Panda M, Purohit P, Wang Y, Meher BR (2022) Functionalized carbon nanotubes as an alternative to traditional anti-HIV-1 protease inhibitors: an understanding towards nano-medicine development through MD simulations. J Mol Graph Model 117:108280. https://doi.org/10.1016/j.jmgm.2022.108280
Purohit P, Dash JJ, Muya JT, Meher BR (2022) Molecular insights to the binding interactions of APNS containing HIV-protease inhibitors against SARS-CoV-2 M(pro): an in silico approach towards drug repurposing. J Biomol Struct Dyn. 1–14. https://doi.org/10.1080/07391102.2022.2059008
Onufriev A, Bashford D, Case DA (2000) Modification of the generalized Born model suitable for macromolecules. J Phys Chem B 104:3712–3720
Roe D, Cheatham T (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular. Dynamics Trajectory Data. 9
Lazarova M (2008) Virtual screening – models, methods and software systems. International Scientific Conference Computer Science. 55–60
Bharadwaj S, Lee KE, Dwivedi VD, Yadava U, Panwar A, Lucas SJ et al (2019) Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against dengue virus NS2B-NS3 protease. Sci Rep 9:19059. https://doi.org/10.1038/s41598-019-55723-5
Rahman MM, Biswas S, Islam KJ, Paul AS, Mahato SK, Ali MA et al (2021) Antiviral phytochemicals as potent inhibitors against NS3 protease of dengue virus. Comput Biol Med 134:104492. https://doi.org/10.1016/j.compbiomed.2021.104492
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51:2778–2786. https://doi.org/10.1021/ci200227u
Acknowledgements
The authors sincerely acknowledge and thank Berhampur University for providing the infrastructure to study this work. All the authors thank the World Bank’s Odisha Higher Education Program for Excellence and Equity (OHEPEE), Department of Higher Education, Government of Odisha, which supports the Centre of Excellence on Bioprospecting of Ethno-pharmaceuticals of Southern Odisha, Berhampur University.
Funding
This work was supported by the Science & Technology (S & T) Department, Government of Odisha, India, with the project (SCST-MISC-0061–2018-1288) to B. R. M. B. R. M. is also supported through UGC, New Delhi, Government of India, with the UGC-BSR Start-Up grant (F.30–484/2019(BSR)). P. P. is also supported through Biju Pattnaik Research Fellowship (BPRF) from S&T, Government of Odisha, India (STBTMISC-0007–2020-2047/ST).
Author information
Authors and Affiliations
Contributions
B. R. M. and P. P. conceived and designed the experiments. P. P., S. S. and B. R. M. performed the experiments. P. P., S. S., M. P., P. S. S. and B. R. M. analysed the data and contributed reagents/materials/analysis tools. P. P. and B. R. M. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Purohit, P., Sahoo, S., Panda, M. et al. Targeting the DENV NS2B-NS3 protease with active antiviral phytocompounds: structure-based virtual screening, molecular docking and molecular dynamics simulation studies. J Mol Model 28, 365 (2022). https://doi.org/10.1007/s00894-022-05355-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05355-w