
Abstract
Usher syndrome, the most prevalent cause of combined hereditary vision and hearing impairment, is clinically and genetically heterogeneous. Moreover, several conditions with phenotypes overlap** Usher syndrome have been described. This makes the molecular diagnosis of hereditary deaf–blindness challenging. Here, we performed exome sequencing and analysis on 7 Mexican and 52 Iranian probands with combined retinal degeneration and hearing impairment (without intellectual disability). Clinical assessment involved ophthalmological examination and hearing loss questionnaire. Usher syndrome, most frequently due to biallelic variants in MYO7A (USH1B in 16 probands), USH2A (17 probands), and ADGRV1 (USH2C in 7 probands), was diagnosed in 44 of 59 (75%) unrelated probands. Almost half of the identified variants were novel. Nine of 59 (15%) probands displayed other genetic entities with dual sensory impairment, including Alström syndrome (3 patients), cone-rod dystrophy and hearing loss 1 (2 probands), and Heimler syndrome (1 patient). Unexpected findings included one proband each with Scheie syndrome, coenzyme Q10 deficiency, and pseudoxanthoma elasticum. In four probands, including three Usher cases, dual sensory impairment was either modified/aggravated or caused by variants in distinct genes associated with retinal degeneration and/or hearing loss. The overall diagnostic yield of whole exome analysis in our deaf–blind cohort was 92%. Two (3%) probands were partially solved and only 3 (5%) remained without any molecular diagnosis. In many cases, the molecular diagnosis is important to guide genetic counseling, to support prognostic outcomes and decisions with currently available and evolving treatment modalities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuro-sensory deficits are among the most prevalent congenital disorders in humans. Impaired hearing and/or vision can negatively affect a person's communication abilities, cognitive functions, and social competencies. Both senses work together and to some extent, one can help compensate for loss of the other. Therefore, deaf–blindness, including a wide range of hearing and vision levels (99% of patients have some residual hearing and/or vision capabilities), is more than the sum of hearing impairment (HI) and vision impairment (VI); it seriously impacts multiple areas of development. Children with dual sensory impairments have specific educational needs, requiring an interdisciplinary team of medical specialists and teachers (Dammeyer 2014).
Deaf–blindness may be acquired, e.g. by intrauterine infections (e.g. rubella, cytomegalovirus), premature birth, and cerebral palsy. However, in developed countries, the vast majority of congenital cases have a genetic basis. Usher syndrome (USH), a heterogeneous autosomal recessive disorder, accounts for approximately 50% of hereditary cases of combined deafness and blindness; it has an incidence of 1 in 6000–25,000 (Kimberling et al. 2010). In addition, variants in USH genes account for up to 10% of children diagnosed with non-syndromic HI (Jouret et al. 2019). The most severe form, USH1, is characterized by congenital severe to profound deafness, vestibular dysfunction, and retinal degeneration in the first decade of life. USH1 can be caused by variants in at least five genes (MYO7A, USH1C, CDH23, PCDH15, and USH1G). The most common form is USH2, accounting for diagnoses in two-thirds of USH patients, that is characterized by moderate to severe congenital HI and retinitis pigmentosa (RP) in the second decade of life. USH2 results from mutations in at least three genes (USH2A, ADGRV1, and WHRN); mutations in USH2A are by far the most common cause accounting for 90% of USH2 cases (Bonnet et al. 2016). The mildest form, USH3, exhibits variable, usually progressive, HI and RP later in life and has been associated with variants in CLRN1 (Hereditary Hearing Loss Homepage; https://hereditaryhearingloss.org; Retinal Information Network; https://sph.uth.edu/retnet/disease.htm).
Here, we performed whole exome analysis (WEA) of 59 unrelated, mainly consanguineous families with at least one proband clinically diagnosed with HI and retinal degeneration. Our study highlights the enormous molecular genetic heterogeneity of combined HI and VI and the need to analyze larger gene panels (for both diseases of the auditory and visual system) in addition to known or suspected USH genes.
Materials and methods
Patients
This study was approved by the ethics committees at the Medical Faculty of Würzburg University, Germany (approval number 46/15), the National Institute of Rehabilitation Luis Guillermo Ibarra (INR), Mexico (no. 12/13), and the Shahid Beheshti University of Medical Sciences, Tehran, Iran and was carried out following the ethical principles of the declaration of Helsinki. Written informed consent was obtained from all participating families prior to their inclusion in the study.
We recruited 59 unrelated probands with combined HI and retinal degeneration resulting in reduced vision and nyctalopia. None of the probands reportedly suffered from intellectual disability. Fifty-two Iranian index patients from consanguineous families were seen by ophthalmologists in different centers and were cataloged in the Iranian Inherited Retinal Disease Registry (NCT04131400) (Sabbaghi et al. 2020). Seven non-consanguineous Mexican probands were recruited by medical geneticists in the Asociación Para Evitar la Ceguera en México (APEC) and the National Institute of Rehabilitation (INR), Mexico City.
Medical, drug and familial histories were recorded. Ocular examinations (Supplementary Table 1) of all Iranian patients were performed at Labbafinejad Medical Center, affiliated with the Shahid Beheshti University of Medical Sciences. These included examination of the best corrected visual acuity (VA) using Snellen charts, slit lamp biomicroscopy of the anterior and posterior segments (using + 90 lens), Goldmann tonometry, and color vision and visual field testing using a Humphrey Field Analyzer II, model 750 (Zeiss Humphrey Systems, Dublin, CA, USA). For patients with VA scores under 20/200, a semi-quantitative scale was used, including counting fingers at different distances, hand movements and light perception. In many cases, depending on the level of vision, standard full field electroretinography (ERG) was performed under photopic and scotopic conditions. Ocular imaging included color fundus photography (CFP), spectral domain optical coherence tomography (OCT) (6 mm scans centered from the fovea, using a Spectralis imaging platform, Heidelberg, Germany), and fundus autofluorescence (FAF) imaging (HRA, Heidelberg, Germany). Patients with apparent USH were classified with mild, moderate or severe cystoid macular edema (CME), using the grading system of Sliesoraityte et al. (2015). The following features were considered: subretinal fluid without clearly detectable cystic macular lesion (CML) boundaries, central macular thickness, largest diameter of CML, calculated mean of all detectable CMLs, total number of detectable CML, and retinal layers affected by CML.
HI was determined by anamnesis and by questionnaire. Audiograms were available from probands 1, 6, 8, 9, 12, 24, 25, 42, 44, 47, 52, 54, and 58 (Supplementary Table 1). Severity of HI was assessed by averaging pure-tone thresholds of the better hearing ear over 0.5, 1, 2, and 4 kHz. Averaged thresholds between 41–70 dB represent moderate, 71–95 dB severe, and > 95 dB profound HL. Interpretation of audiometry followed the recommendations of the GENDEAF study group (Mazzoli et al. 2003).
Sequence analysis and variant classification
Genomic DNAs of probands and available family members were extracted from whole blood samples using a standard salting out method and quantified using Qubit 2.0 (Life Technologies, Carlsbad, CA, USA). Exome capture was performed using the TruSeq Rapid Exome or Nextera DNA Exome (Illumina, San Diego, CA, USA) enrichment according to manufacturer’s protocols. Libraries were paired-end sequenced (2 × 76 bp) with the v2 reagent kit (Illumina) on a NextSeq 500 (Illumina) sequencer. The generated sequences were de-multiplexed and mapped to the human genome reference (NCBI build 37/hg19 version) with Burrows Wheeler Aligner.
First, a targeted analysis of the following USH-associated genes was performed using GensearchNGS software (PhenoSystems SA, Wallonia, Belgium): ABDH12 (OMIM 613599, NM_001042472.2), ADGRV1 (OMIM 602851, NM_032119.3), CDH23 (OMIM 605516, NM_022124.5), CEP250 (OMIM 609689, NM_007186.5), CLRN1 (OMIM 606397, NM_001195794.1), MYO7A (OMIM 276903, NM_000260.3), PCDH15 (OMIM 605514, NM_001142763.1), PDZD7 (OMIM 612971, NM_001195263.1), USH1C (OMIM 605242, NM_153676.3), USH1G (OMIM 607696, NM_173477.4), USH2A (OMIM 608400, NM_206933.2), and WHRN (OMIM 607928, NM_015404.3). In a second step, the whole exome was analyzed in patients without molecular USH diagnoses. A refuted USH1J gene, CIB2 (Riazuddin et al. 2012; Booth et al. 2018), a questionable USH1M gene, ESPN (Ahmed et al. 2018), a refuted USH3B gene, HARS (Puffenberger et al. 2012; DiStefano et al. 2019), and a recently identified USH4 gene, ARSG (Peter et al. 2021), were not included in our original screen, but rather in exome-wide analysis.
Single nucleotide variants (SNVs) and small indels (< 15 bp) were analyzed using GensearchNGS, MutationDistiller (Hombach et al. 2019), and Moon Diploid (http://www.diploid.com/moon). Alternative alleles present at > 20% and a minor allele frequency (MAF) < 0.01 were used for variant filtering. Population-specific allele frequencies were assessed using gnomAD (Karczewski et al. 2020) and the Greater Middle Eastern (GME) variome (Scott et al. 2016). PolyPhen-2 (Adzhubei et al. 2010), MutationTaster (Ng and Henikoff 2001), and SIFT (Schwarz et al. 2014) were used to analyze pathogenicity of SNVs. Variants were queried in the Deafness Variation Database (DVD) (Azaiez et al. 2018), the Human Gene Mutation Database (HGMD) (Stenson et al. 2014), and the Leiden Open Variation Database (LOVD) (https://www.lovd.nl). Potential splicing effects of variants were classified by in silico prediction tools such as SpliceSiteFinder-like (Shapiro and Senapathy 1987), MaxEntScan (Yeo and Burge 2004), NNSPLICE (Reese et al. 1997), Genesplicer (Pertea et al. 2001), and Human Splicing Finder (Desmet et al. 2009). Variants were classified according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (Richards et al. 2015), as well as established recommendations for hereditary HI (Oza et al. 2018). Potentially disease-causing and compound heterozygous variants were validated by segregation analysis in the index patients and available family members using Sanger sequencing on an ABI 3130xl 16-capillary sequencer (Life Technologies). Primer sequences were designed using Primer 3 (https://primer3.org).
In probands with a single pathogenic USH2A variant (50, 51, and 57), partially solved (45 and 53), and unsolved cases (55, 57, and 59), multiplex ligation-dependent probe amplification (MLPA) with the SALSA Probemix P361, P362, and P292 (MRC Holland, Amsterdam, The Netherlands) was used to exclude copy number variations (CNVs) in USH2A and PCDH15. In addition, these probands were screened by Sanger sequencing for deep intronic mutations (DIMs) in USH2A introns 27 (c.5573-843A>G), 40 (c.7595-2144A>G), 44 (c.8845 + 628C>T), 50 (c.9959-4159A>G), and 64 (c.14134-3169A>G) (Vache et al. 2012; Liquori et al. 2016; Baux et al. 2017). In some probands showing multi-locus variation (42, 43, 44, 45, and 58), the Infinium Global Screening Array-24 v1.0 BeadChip (Illumina, SanDiego, CA, USA) was applied for genome-wide CNV screening.
In vitro splice assays
To evaluate the impact of the ADGRV1 c.9623 + 3A>C and PDSS2 c.702 + 1G>A splice variants, PCR amplification from patient and control genomic DNA ensued following a modified protocol (Tompson and Young 2017). For testing of the ADGRV1 variant, amplification of a region including exon 44 (176 bp) with an additional 589 bp (5′) and 340 bp (3′) from the flanking intronic region was performed using primers containing XhoI and BamHI restriction sites (forward primer with a XhoI restriction site: 5′-aattctcgagTTTGCTGGTTCTTGAGCTTC-3′ and reverse primer with a BamHI restriction site: 5′-attggatccTCATTCACTTGGTTTGAGCAG-3′). Similarly, for testing of the PDSS2 variant, amplification of a region containing exon 4 (71 bp) that included 390 bp (5′) and 1380 bp (3′) from the flanking intronic regions was performed (forward primer with a XhoI restriction site: 5′-aattctcgagTTGTAATTGCTCCCAGAATGG-3′ and reverse primer with a BamHI restriction site: 5′-attggatccTGACTTCAAATCCCTGAGAGC-3′). Following PCR amplification and clean-up, restriction enzyme digestion of the PCR fragment and pSPL3 exon trap** vector was performed prior to ligation between exons A and B of the linearized pSPL3-vector. The vector was transformed into DH5α competent cells (NEB 5-alpha, New England Biolabs, Frankfurt am Main, Germany), plated, and incubated overnight. The wild-type and mutant-containing vector sequences were Sanger sequence confirmed. Vectors containing mutant and wild-type sequence were transfected into HEK 293T cells (ATCC) at a density of 2 × 105 cells per milliliter. 1 µg of the respective pSPL3 vectors were transiently transfected using 3 µl of FuGENE 6 Transfection Reagent (Promega, Walldorf, Germany). An empty vector and transfection negative reactions were included as controls. The transfected cells were harvested 24 h after transfection. Total RNA was prepared using miRNAeasy Mini Kit (Qiagen, Hilden, Germany). Approximately 1 µg of RNA was reverse transcribed using a High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s protocols. The cDNA was PCR amplified using vector-specific SD6 forward (5′-TCTGAGTCACCTGGACAACC-3′) and SA2 reverse (5′-ATCTCAGTGGTATTTGTGAGC-3′) primers. The amplified fragments were visualized on a 2% agarose gel and Sanger sequenced. ADGRV1 fragments were cloned using the TA cloning dual promoter with pCRII kit (Invitrogen, Karlsruhe, Germany) and subsequently Sanger sequenced.
Results
Clinical and molecular characterization of the analyzed cohort
Staged exome diagnostics was performed on 59 unrelated probands with co-occurring HI and VI (due to retinal dystrophy) and without apparent intellectual disability. Patient age at the time of recruitment ranged from 8 to 65 years with a mean age of 34 years. Clinical parameters are presented in Supplementary Table 1. The reported visual acuity ranged from 20/30 Snellen equivalent to no light perception. The most common symptom was nyctalopia accompanied by bilateral reduced vision. The most common ophthalmological findings included waxy pallor discs, retinal atrophy, mottling and diffuse bone spicule pigmentary changes, cataracts, and macular edema. With exception of proband 11, intraocular pressure of both eyes by Goldmann tonometry was normal. The majority (64%) of the probands presented with prelingual HI, the rest with postlingual HI. If available, tympanograms showed normal middle ear function.
Forty-seven of the 52 (90%) cases from consanguineous Iranian families and all 7 cases from non-consanguineous Mexican families were solved. The overall diagnostic yield was 92% (54 of 59). A molecular diagnosis pointing to USH accounted for 44 of 59 (75%) and non-USH syndromes for 9 of 59 (15%) index probands. In three of the USH cases, additional variants in genes for non-syndromic HI (OTOG, TECTA) or ABCA4-related VI (Stargardt disease, cone-rod dystrophy, and RP) may modify/aggravate the phenotype (Fig. 1, left). In one of 59 (2%) probands, HI and VI were caused by variants in different genes. Two additional probands with a blended phenotype were partially solved.

The left diagram shows the diagnostic yield in 59 probands with combined vision and hearing impairment. As expected, USH was the most common diagnosis accounting for 75% of cases. Please note that three USH (+ multi-locus variation) probands exhibited variants in additional genes for HI and VI. Different non-USH syndromes with co-occurring HI and VI were found in 15% of cases. The right diagram represents the mutational spectrum in the 44 USH probands. Mutations in USH1 genes are marked in different shades of blue and in USH2 genes in green
Usher syndrome as the most common cause of dual sensory loss
Forty-four patients showed biallelic variants in known USH genes, approximately half of the variants being novel (Table 1; Fig. 1, right). Nineteen of these 44 patients suffered from USH1, 24 from USH2, and only one from USH3. The most frequently affected genes in our cohort were MYO7A (USH1B) in 16 patients and USH2A (USH2A) in 17 patients, followed by ADGRV1 (USH2C) in 7 patients. Biallelic variants in USH1C, CDH23 (USH1D), USH1G, and CLRN1 (USH3A) were diagnosed in single patients each. It is noteworthy that although the vast majority of our USH probands were from consanguineous families, compound heterozygous variants were found in 18 of 44 (41%) cases. Fundus examination, FAF, and OCT of selected patients with USH showed findings consistent with RP (Fig. 2). Using a grading system for CME (Sliesoraityte et al. 2015), moderate and severe cystic lesions were most prevalent among patients with USH1B and biallelic MYO7A variants (Supplementary Table 1). Bilateral pantonal profound sensorineural HI without vestibular dysfunction was the most common form of HI in the USH cohort (Supplementary Table 1).

Retinal phenotype of selected probands with co-occurring HI and VI due to USH entities consistent with retinitis pigmentosa. A, D, G, J, M, P Fundus photography (FP), B, E, H, K, N, Q fundus autofluorescence (FAF), and C, F, I, L, O, R optical coherence tomography (OCT) imaging of patients 1 (A–C) and 7 (D–F) with USH1B, patient 18 (G–I) with USH1C, patient 41 (J–L) with USH1D, patient 21 (M–O) with USH2A, and patient 37 (P–R) with USH3A. Note retinal atrophy along the arcades in all patients, and bone spicules in patient 21 on FP imaging, decreased autofluorescence along the arcades, and increased and/or decreased autofluorescence centrally in all patients on FAF imaging, and macular pathology on OCT imaging in all patients, i.e. atrophy in patient 1, cystoid macular edema in patients 1, 7, 41, 21, and 37, and epiretinal membrane in patient 18
Patient 33 displayed a homozygous likely pathogenic splice variant, ADGRV1 c.9623 + 3A>C (Table 1). In vitro splice assays showed three bands after RT-PCR (Supplementary Fig. 1). TA cloning showed 54% of transcripts using a first cryptic splice site 129 nucleotides before the native exon 44 splice donor site, leading to an in-frame deletion, r.9495_9623del p.(Tyr3166_Arg3208del); 38% of transcripts where splicing involved the second cryptic splice donor site 94 nucleotides before the exon 44 native splice site, causing a frameshift, r.9530_9623del p.(Gly3177Glufs*5), and 8% of misspliced transcripts showed skip** of exon 44, r.9448_9623del p.(Ala3150Serfs*11), also resulting in a frameshift. The wild-type minigene showed two main bands comprised of an upper band containing the expected exon 44 sequence (433 bp) and a lower band showing a skipped exon 44 (257 bp) that maps to exon 1 of a shorter transcript (ENST00000640464.1) lacking a splice acceptor site for splicing.
No pathogenic variants in CIB2 (USH1J), ESPN (USH1M), HARS (USH3B), and ARSG (USH4) were found by WEA in the remaining 15 non-USH probands. Probands 50, 51, and 57 who exhibited single variants in USH2A, were analyzed by MLPA and Sanger sequencing to exclude CNVs and DIMs, which are not detectable by WEA. In addition to a novel pathogenic variant, c.5438C>A p.(Ser1813*), proband 43 was endowed with a well-known DIM, c.7595-2144A>G, in exon 40 (Table 1). Since her unaffected sister displayed only the pathogenic nonsense variant but not the DIM, this is consistent with the diagnosis USH2A in the index patient. Moreover, proband 43 (but not her unaffected sister) displayed a variant of uncertain significance (VUS) in TECTA (Table 1), which is associated with autosomal dominant deafness (DFNA8/12) and may contribute to her HI.
Proband 42, a 25-year-old female, with bilateral severe prelingual HI, bilateral reduced vision and nyctalopia starting at the age of 7 years, macular atrophy, diffuse retinal atrophy and pigmentary changes displayed a homozygous splice mutation, USH2A c.11389 + 3A>T (Table 1), which has been associated with retinal dystrophy and USH2A. An in vitro minigene assay has been reported, confirming truncation of 48 bp due to usage of a cryptic donor splice site in exon 58 (Soens et al. 2017). In addition, proband 42 was endowed with a homozygous missense variant, c.7454del p.(Arg2485Hisfs*77) in OTOG (Table 1), which causes autosomal recessive deafness (DFNB18B; OMIM 614945) (Downie et al. 2020) and may aggravate the HI phenotype. Her father and two unaffected siblings were heterozygous for this variant. The VI of proband 42 may be modified by the heterozygous variant c.632G>A p.(Arg211Gln) in PRPF31 (Table 1), which has been associated with RP11 (OMIM 600138) in a single sporadic RP case (Rose et al. 2016). Although the MAF of this variant is about 0.001, functional studies have associated incomplete penetrance and variable expressivity of RP11 with PRPF31 mRNA levels (Rivolta et al. 2006; Rio Frio et al. 2009). Moreover, proband 42 displayed a heterozygous VUS, c.632G>A p.(Arg287Trp) in ROM1 (Table 1), which can cause RP7 together with mutations in PRPH2 (Kajiwara et al. 1994). In this example of digenic RP, ROM1 is considered as a dominant modifier of PRPH2 (Nadeau 2001).
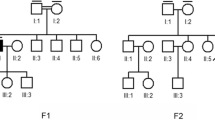
Proband 52, a 48-year-old female with prelingual onset, bilateral, profound, steeply slo** high-frequency HI with type A tympanogram and bilateral reduced vision, nyctalopia, and photophobia starting at 5 years of age, showed two missense variants in ADGRV1, c.2261T>C p.(Val754Ala) and c.10878A>C p.(Lys3626Asn) (Table 1). Since two unaffected siblings were each heterozygous for either variant, the proband must be compound heterozygous, consistent with the diagnosis USH2C (Fig. 3B, C). The early onset and rapidly progressing vision loss (with light perception in the left eye and no light perception in the right eye), which is incompatible with USH2C, may be explained by an additional homozygous variant c.4919G>A p.(Arg1640Gln) in ABCA4 (Table 1), which causes Stargardt disease (Simonelli et al. 2000). The most recent ocular examination revealed bilateral pseudophakic status, pale discs, atrophy and pigmentary changes in the macula, atrophy, bone spicules and pigment clumps in the retinal periphery (Fig. 3A). The variants segregated in a heterozygous state in two unaffected brothers (Fig. 3B, C).

Possible coincidence and genetic heterogeneity underlying combined HI and VI. A Fundus photography (FP) of proband 52 showing central retinal atrophy and pigment clum** in both eyes consistent with ABCA4-spectrum disease. B, C Pedigree and segregation analysis of disease-causing variants in ABCA4 and ADGRV1. D FP, fundus autofluorescence (FAF) and optical coherence tomography (OCT) imaging of proband 44 showing decreased autofluorescence along the inferior arcade and centrally on FAF imaging (right eye) and retinal atrophy on OCT imaging consistent with retinal dystrophy. E, F Pedigree and segregation analysis of variants in MYH14 and FBN2. (+/+ variant present in homozygous state, ± variant present in heterozygous state, −/− wild type.)
In addition to a homozygous frameshift mutation in USH2A, c.236_239dup p.(Gln81Tyrfs*28), proband 58 presented with a medically actionable pathogenic variant, c.733_734del p.(Gly245Argfs*39) in KCNQ1 (Table 1) (Amirian et al. 2018), which is associated with long QT syndrome 1 (OMIM 192500) and familial atrial fibrillation 3 (OMIM 607554).
Non-USH syndromes causing combined HI and retinal degeneration
Nine of 59 probands with combined HI and retinal degeneration displayed clinical entities different from USH (Table 2). These included well-known syndromes that should be considered as part of the differential diagnosis of USH as well as unexpected inborn errors of metabolism. In many of the identified cases, the age-dependent manifestation of additional symptoms and the large clinical heterogeneity rendered a clinical diagnosis difficult or even impossible, hereby supporting the implementation of genetic testing.
It is noteworthy that three probands (47, 48, and 49) were endowed with different homozygous variants, c.6299C>A p.(Ser2100*), c.7471_7472del p.(Ser2491Thrfs*5), and c.11410C>T p.(Arg3804*), respectively, in ALMS1, causing Alström syndrome (OMIM 203800) (Table 2). This pleiotropic ciliopathy is associated with HI and VI, cardiomyopathy, endocrine, neurological, and hepatic symptoms (Rethanavelu et al. 2020). At the time of recruitment, our three Alström patients presented with photophobia, visual field constriction, posterior subcapsular cataracts, pigmentary retinopathy (Fig. 4A, B), and progressive sensorineural HI without vestibular dysfunction. Proband 47 suffered from bilateral severely reduced vision and bilateral profound steeply slo** high-frequency HI. Ophthalmological examinations revealed normal intraocular pressure, a pseudophakic status of the anterior segment, chalky white pale discs, severe macular atrophy (beaten bronze), diffuse bone spicule pigmentary changes, and mottling. At the time of examination, no clinical sign of cardiomyopathy was seen in our three Alström probands. Retrospectively, we learnt that proband 47 suffered from insulin-resistant diabetes and renal failure.

Retinal phenotype of selected probands with co-occurring HI and VI due to non-USH syndromic entities. A, B Fundus photography (FP) and fundus autofluorescence (FAF) in proband 49 with Alström syndrome (ALSM1) showing bone spicules in the retinal periphery (right and left eye) consistent with retinitis pigmentosa (RP). C, D FP and FAF of proband 46 with Heimler syndrome (PEX26) showing optic atrophy and pigment clum** despite reduced image quality due to cataracts (both eyes) consistent with RP. E–H FP, FAF, and optical coherence tomography (OCT) imaging of proband 39 with CRDHL1 (CEP78) showing decreased autofluorescence along the arcades on FAF imaging (right eye) and atrophy of the outer retina outside the center on OCT imaging (both eyes) consistent with RP. I–L FP, FAF, and OCT of proband 50 with Scheie syndrome (IDUA) showing decreased autofluorescence along the arcades on FAF imaging (right eye) and atrophy of the outer retina outside the center and mild cystoid macular edema on OCT imaging (both eyes) consistent with RP
Two patients, 39 and 40, suffered from another ciliopathy, namely cone-rod dystrophy and hearing loss 1 (CRDHL1; OMIM 617236), due to homozygous or compound heterozygous variants in CEP78, respectively (Table 2). A possible founder mutation, c.515T>G p.(Ile172Arg) was identified. This variant has not been annotated in gnomAD, GME, LOVD, and HGMD. It was homozygous in a 32-year-old female (proband 39) who reported HI starting at the age of 5 years and bilateral reduced vision, photophobia and hemeralopia at 26 years. Clinical examination revealed pale discs, mild macular atrophy, diffuse retinal atrophy, and retinal pigment epithelium (RPE) mottling in both eyes (Fig. 4E–H). Kinetic and/or static visual field examination revealed a generalized depression with reduced mean deviation (− 3.344 dB; p < 0.01) with a central 10–2 threshold, consistent with RP. The ERG showed severe reduced amplitude in scotopic, photopic, and flicker responses.
A novel homozygous missense VUS in PEX26, c.349C>A p.(Pro117Thr) (Table 2) was found in a 12-year-old female (proband 46) with bilateral reduced vision, nyctalopia, bilateral subcapsular cataract, diffuse retinal atrophy with diffuse pigmentary changes in both eyes (Fig. 4C, D), and severely reduced amplitudes in scotopic and photopic ERG. This variant segregated in a heterozygous state in unaffected family members who were available for testing. Pathogenic variants in PEX26 are associated with a wide spectrum of peroxisomal diseases, including Heimler syndrome, previously described in patients with concurrent HI and VI (Neuhaus et al. 2017). When the molecular diagnosis was communicated to the recruiting clinic, amelogenesis imperfecta in secondary teeth was retrospectively identified, which is in line with Heimler syndrome.
The homozygous likely pathogenic variant c.956C>T p.(Ala319Val) in IDUA in proband 50 (Table 2) has been previously reported in patients with Scheie syndrome (OMIM 607016), a mild form of mucopolysaccharidosis type I (Beesley et al. 2001). The 65-year-old female patient presented with diffuse RPE mottling, CME, generalized depression in her visual field exam (Fig. 4I–L), and postlingual HI. There were no other evident clinical findings indicative of Scheie syndrome.
The 29-year-old proband 51 with optic atrophy, retinal degeneration, and sensorineural HI was compound heterozygous for a missense variant c.488G>A p.(Arg163His) and a splice site variant c.702 + 1G>A p.? in PDSS2 (Table 2) that has been associated with coenzyme Q10 deficiency (OMIM 614652). Compound heterozygosity of the respective VUS and likely pathogenic splice variant was confirmed by segregation analysis in unaffected family members. In vitro splice testing of the PDSS2 c.702 + 1G>A mutant allele indicated skip** of exon 4 (257 bp), leading to an in-frame deletion, r.631_702del p.(Val211_Lys234del). The wild-type amplicon included the correctly spliced complete exon 4 sequence, showing as a 328 bp band (Supplementary Fig. 1).
The homozygous variant c.1171A>G p.(Arg391Gly) in ABCC6 (Table 2) has been previously associated with pseudoxanthoma elasticum (Chassaing et al. 2004). Proband 54, a 31-year-old female, presented with progressive visual impairment, optic disc drusen, beaten bronze macular atrophy, diffuse RPE mottling, choroidal neovascularization, and postlingual sensorineural HI.
Possible multi-locus genomic variation
Proband 44, a 42-year-old female, suffered from bilateral moderate high-frequency HI as well as late-manifesting (at the age of 38 years) severely reduced visual acuity, photophobia, nyctalopia, severe macular atrophy, diffuse pigmentary changes, and mottling with notable diminished central macular thickness (Fig. 3D). She displayed three novel missense variants; one, c.4732A>G p.(Lys1578Glu) in MYH14, and two, c.7355A>C p.(Glu2452Ala) and c.2507C>T p.(Thr836Met) in FBN2 (Table 3). Since the unaffected brother did not exhibit FBN2 variants (Fig. 3E, F) and other family members were not available, we do not know whether both variants reside on the same or on different parental alleles. Rare dominant variants in FBN2 have been described in relationship with early onset macular degeneration (OMIM 616118) but also age-related macular degeneration (Ratnapriya et al. 2014). Variants in MYH14 cause autosomal dominant HI (DFNA4A; OMIM 600652).
Proband 45, a 40 year-old male, reported reduced vision and nyctalopia starting at 10 years of age and bilateral postlingual HI. He displayed macular pathology with pigmentary changes and bone spicules in the retinal periphery. He comes from a large consanguineous family. There are multiple members with dual sensory impairment and one brother with isolated HI. Both the proband and his deaf–blind youngest sister were endowed with a missense variant c.659T>G p.(Phe220Cys) in RHO (Table 3), that has been previously reported in patients with RP4 (OMIM 13731) (Bunge et al. 1993). Since the variant was also present in the hearing-impaired brother, reduced penetrance and/or variable expressivity must be assumed and the evidence for this variant is limited. Functional studies of RP-linked rhodopsin mutations have yielded conflicting results (Mallory et al. 2018; Lewis et al. 2020). The molecular basis of the HI in this family remains unknown. As an incidental finding, the proband displayed a common pathogenic variant in MEFV, that has been associated with familial Mediterranean fever and Behcet's disease (Tasliyurt et al. 2013).
In proband 53, a 13-year-old girl, we identified aa heterozygous variant, c.6462C>A p.(His2154Gln), in PRPF8 (Table 3), which has been associated with RP13 (OMIM 600059), which is typically inherited as an autosomal dominant trait. In addition, she was endowed with a homozygous variant c.2551 + 8C>T in CACN2D4 (Table 3), which has been associated with retinal cone dystrophy 4 (OMIM 610478). Variants in both genes may contribute to VI within the first year of life. A segregating variant underlying the HI has not been identified.
Unsolved cases
In 3 of 59 probands (55, 57, and 59), we could not establish a molecular diagnosis for HI and/or VI. Interestingly, proband 57 exhibited a single pathogenic mutation, c.5388T>A p.(Cys1796*), in USH2A and proband 59 was heterozygous for two VUS, c.3750 + 7G>A p.? in MYO7A and c.5653C>T p.(Arg1885Cys) in CDH23 (Table 3). Proband 59, a 47-year-old male with prelingual HI and VI since birth, was also endowed with a pathogenic variant, c.1057C>T p.(Pro353Ser) in G6PD (Beutler et al. 1992), which is associated with X-linked hemolytic anemia (OMIM 300908) in hemizygous males. Hyperbilirubinemia due to G6PD deficiency can induce neurological damage (kernicterus) in neonates and children, which may clinically manifest as auditory neuropathy spectrum disorder (Boskabadi et al. 2018).
Discussion
As expected, most (44 of 59; 75%) of our deaf–blind patients were diagnosed with USH. Approximately half were USH1 and half USH2, with MYO7A (USH1B) and USH2A being the most prevalent genes. The relatively high proportion of ADGRV1 (USH2C) (7 of 24; 29%) compared to USH2A (17 of 24; 71%) within the USH2 group may be explained by ethnicity of our cohort, mainly from Iran, which is an understudied population. In addition to USH, there is a wide variety of hereditary, non-hereditary and independent causes of HI and/or VI, which makes a correct diagnosis for clinicians and human geneticists challenging (Stiff et al. 2020).
Next generation sequencing (NGS) has greatly transformed the molecular diagnostics of both clinically and genetically highly heterogeneous neuro-sensory disorders, allowing rapid screening of large gene panels or the entire exome. Using WEA, we found genetic variants associated with combined HI and VI in 54 of 59 (92%) probands. Two probands (3%) were partially solved and only three (5%) remained without a molecular diagnosis explaining their phenotype. This is consistent with recent NGS screens of patients with combined HI and VI, which have yielded diagnostic efficiencies > 90% (Bonnet et al. 2016; Neuhaus et al. 2017; Jouret et al. 2019). This unusually high solve rate for a heterogeneous Mendelian disorder argues in favor of the notion that most genes underlying dual sensory loss have been identified. Our study also shows the utility of OCT for monitoring RP patients. The presence of macular edema, macular hole, epiretinal membrane, etc. has prognostic value and therapeutic implications.
The vast majority of variants underlying USH in our deaf–blind cohort were classified as pathogenic (44 of 63; 70%) or likely pathogenic (11 of 63; 18%) and most of them were already known (36 of 63; 57%). In contrast, the majority of presumably disease-causing variants in non-USH probands were VUS (10 of 17; 59%) and/or novel (12 of 17; 71%). This is not unexpected, considering that USH is one of the most extensively studied neuro-sensory disorders and updated comprehensive information on USH genes is available in variant data bases. Compared to USH, the data situation of the genes identified in probands with non-USH syndromes and multi-locus variation is relatively poor. Therefore, segregation analyses were performed in the families of the index probands to validate or discard variants.
Non-Usher syndromes associated with combined hearing loss and retinal degeneration
Although USH is by far the most prevalent cause of deaf–blindness worldwide and also in our study, there are other syndromes that combine retinal dystrophy and HI with a number of additional symptoms. Five of our probands exhibited ciliopathies due to variants in ALMS1 (Alström syndrome) and CEP78 (CRDHL1). The absence of additional clinical symptoms in our three Alström patients may be explained by the variable and age-dependent expressivity of the phenotype. Only 17 and 19% of patients with Alström syndrome display intellectual disability and cardiomyopathy, respectively (Rethanavelu et al. 2020).
Heimler syndrome is a peroxisome disorder caused by biallelic variants in PEX1, PEX6, and PEX26. Proband 46 and several published cases with suspected USH presented biallelic mutations in PEX1 and PEX26 (Neuhaus et al. 2017; Diñeiro et al. 2020). The typical tooth (enamel) and nail abnormalities of Heimler syndrome were overlooked before molecular diagnosis. When assessed retrospectively, our proband presented mild amelogenesis imperfecta in secondary teeth.
Although it is known that patients with alpha-l-iduronidase deficiency (MPS1S or Scheie syndrome) can have HI and retinal degeneration, it was surprising that these were the cardinal symptoms leading to a molecular diagnosis (homozygous IDUA variant) in proband 50. This illustrates that mild phenotypic expression, in this case of a MPS1S, can complicate or delay diagnosis.
Another unexpected finding was compound heterozygosity of a likely pathogenic variant and a novel missense variant in PDSS2 in proband 51. Coenzyme Q10 (CoQ10) deficiency is characterized by highly variable multi-systemic manifestations, ranging from fatal neonatal encephalopathy with hypotonia to isolated steroid-resistant nephrotic syndrome. The establishment of this molecular diagnosis in one of our probands with HI and VI is especially relevant, since high-dose oral CoQ10 supplementation can slow disease progression and even reverse some manifestations (Alcazar-Fabra et al. 2018).
Proband 54 was homozygous for a known variant in ABCC6, causing pseudoxantoma elasticum (OMIM 264800). This systemic elastic tissue disorder progressively affects the skin, forming yellowish papules that coalesce to form plaques until the skin becomes loose and redundant. Ophthalmological findings include angioid streaks, reticular macular dystrophy and speckled appearance of the macula. The clinical diagnosis is typically made in the second or third decade of life, when the skin and retinal symptoms are evident. Although HI has not been associated with pseudoxanthoma elasticum, it has been reported in patients with generalized arterial calcification of infancy (OMIM 624473), which is also caused by recessive variants in ABCC6.
Collectively, these results suggest that patients with dual sensory loss as the primary symptoms can suffer from a long list of syndromes (Stiff et al. 2020) that have HI and VI as part of their symptoms and with mild phenotypic expression or absence of additional symptoms that define the syndrome. In many monogenic disorders, the genotype is not predictive of the phenotype (Cooper et al. 2013). Variants that have been found in patients with highly variable phenotypic manifestations and apparently normal healthy individuals underscore the importance of variable expressivity and reduced penetrance.
Blended phenotypes
Large WEA studies revealed dual molecular diagnoses in a considerable number of patients (Yang et al. 2014; Balci et al. 2017; Posey et al. 2017). Therefore, it is not surprising that three of our Usher probands exhibited additional variants in genes causing HI (OTOG, TECTA) or VI (ABCA4). One non-USH proband exhibited variants in different genes underlying HI (MYH14) and retinal or macular degeneration (FBN2). In two probands, we found variants segregating with VI but no variants in deafness genes. Altogether, in 6 (10%) of 59 probands dual sensory loss may represent a blended phenotype of variants in different genes for HI and/or VI.
In this context, it is noteworthy that although the vast majority of our patients come from consanguineous families, 20 of 54 (37%) solved cases are due to compound heterozygous variants in recessive genes. In 5 of 6 probands with likely multi-locus variation, dominant variants contributed to the phenotype. This accumulation of heterozygous variants in deaf–blind families may be at least partially related to phenotypic mating structure among individuals with HI and/or VI.
Limitations
In probands with a single pathogenic USH2A variant (50, 51, and 57), partially solved (45 and 53), and unsolved cases (55, 57, and 59), CNVs and DIMs in USH2A were excluded by MLPA and Sanger sequencing. Similarly, CNVs in PCDH15 were excluded by MLPA. In some probands (42, 43, 44, 45, and 58), genome-wide microarray screening revealed several CNVs; however, none were associated with HI and/or VI. Although we did not perform a comprehensive CNV analysis in all our probands, we can largely exclude a major contribution of CNVs to the etiopathogenesis of deaf–blindness in our cohort.
Despite diagnostic yields over 90%, some patients with dual sensory impairment remain without firm molecular diagnosis. This may be due to not yet discovered genes or variants in non-coding (intronic and regulatory) sequences, undetected CNVs and structural variants (i.e. inversions), or unannotated exons of known genes. Indeed, two of three unsolved patients displayed single pathogenic variants in USH genes, arguing in favor of the notion that we may have missed the mutation in the second allele. Although switching molecular diagnostics from WEA to whole-genome sequencing and improvement of diagnostic algorithms may overcome some of these problems, our capacity for interpretation variants outside the exome is still very limited.
Benefits of improved molecular diagnostics
Many patients with neuro-sensory impairments benefit from an early molecular diagnosis, which may have important implications for disease management (i.e. tailoring optimum educational programs) and treatment (i.e. cochlear implantation, timely eye examinations and implementation of prophylactic or therapeutic measures to improve vision or slow the progression of retinal degeneration), prognosis (progressive or stable, development of additional symptoms), and family planning. Diagnosing syndromes in patients who are pre-symptomatic (for a given symptom) enables patients to consult respective specialists before symptoms manifest or progress. A variety of gene therapy approaches using adeno-associated viral vectors for gene delivery, antisense oligonucleotides or genome editing agents have already yielded promising results to prevent HI and retinal degeneration in murine USH models (Nagel-Wolfrum et al., 2014; Géléoc and El-Amraoui, 2020; Lentz et al. 2020), including clinical trials in patients related to mutations in exon 13 of USH2A (NCT03780257). Molecular diagnosis of the underlying defect is crucial to stratify patients for a growing number of successful ongoing clinical trials of ocular gene therapy, which represents a notable advancement to other inherited disorders (Cehajic-Kapetanovic et al. 2020).
Using state-of-the art clinical exome analysis, incidental or secondary findings unrelated to the primary reason for sequencing but of medical value for the proband, are identified in several percent of cases (Hart et al. 2019). The ACMG recommends returning highly penetrant pathogenic variants for a list of several dozen genes (Kalia et al. 2017; https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/). Important inclusion criteria are the possibility to confirm the associated phenotype, the availability of preventive measures and treatments, and that the mutation carriers can be asymptomatic for prolonged periods of time. Ideally, the patients should be alerted to this possibility before testing and have the chance to opt-out of receiving such unexpected findings. In this study we detected one medically actionable variant in KCNQ1, which is associated with long QT syndrome and has immediate implications for patient management. In addition, we found a pathogenic variant in MEFV which is associated with familial Mediterranean fever and a hemizygous variant in G6PD, which is associated with X-linked hemolytic anemia. Although not included in the list of actionable genes, these variants were considered as medically relevant and returned to the affected probands/families. Thus, altogether three (5%) probands in our cohort displayed secondary findings.
Web resources
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar.
Deafness Variation Database, https://deafnessvariationdatabase.org.
Hereditary Hearing Loss Homepage, https://hereditaryhearingloss.org.
Leiden Open Variation Database (LOVD), https://www.lovd.nl.
Moon Diploid, https://www.diploid.com/moon.
Mutation Distiller, https://www.mutationdistiller.org.
Primer 3, https://primer3.org/.
ACMG Recommendations for Reporting of Incidental Findings in Clinical Exome and Genome Sequencing, https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/.
Retinal Information Network, https://sph.uth.edu/retnet/disease.htm.
Availability of data and materials
All data generated during this study are included in this article and supplementary information file. All variants have been entered into LOVD. Accession numbers are included in Supplementary Table 2.
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Ahmed ZM, Jaworek TJ, Sarangdhar GN, Zheng L, Gul K, Khan SN, Friedman TB, Sisk RA, Bartles JR, Riazuddin S, Riazuddin S (2018) Inframe deletion of human ESPN is associated with deafness, vestibulopathy and vision impairment. J Med Genet 55:479–488
Alcazar-Fabra M, Trevisson E, Brea-Calvo G (2018) Clinical syndromes associated with coenzyme Q10 deficiency. Essays Biochem 62:377–398
Amirian A, Zafari Z, Dalili M, Saber S, Karimipoor M, Dabbagh Bagheri S, Fazelifar AF, Zeinali S (2018) Detection of a new KCNQ1 frameshift mutation associated with Jervell and Lange-Nielsen syndrome in 2 Iranian families. J Arrhythm 34:286–290
Auslender N, Bandah D, Rizel L, Behar DM, Shohat M, Banin E, Allon-Shalev S, Sharony R, Sharon D, Ben-Yosef T (2008) Four USH2A founder mutations underlie the majority of Usher syndrome type 2 cases among non-Ashkenazi Jews. Genet Test 12:289–294
Azaiez H, Booth KT, Ephraim SS, Crone B, Black-Ziegelbein EA, Marini RJ, Shearer AE, Sloan-Heggen CM, Kolbe D, Casavant T, Schnieders MJ, Nishimura C, Braun T, Smith RJH (2018) Genomic landscape and mutational signatures of deafness-associated genes. Am J Hum Genet 103:484–497
Bademci G, Cengiz FB, Foster Ii J, Duman D, Sennaroglu L, Diaz-Horta O, Atik T, Kirazli T, Olgun L, Alper H, Menendez I, Loclar I, Sennaroglu G, Tokgoz-Yilmaz S, Guo S, Olgun Y, Mahdieh N, Bonyadi M, Bozan N, Ayral A, Ozkinay F, Yildirim-Baylan M, Blanton SH, Tekin M (2016) Variations in multiple syndromic deafness genes mimic non-syndromic hearing loss. Sci Rep 6:31622
Balci TB, Hartley T, ** Y, Dyment DA, Beaulieu CL, Bernier FP, Dupuis L, Horvath GA, Mendoza-Londono R, Prasad C, Richer J, Yang XR, Armour CM, Bareke E, Fernandez BA, McMillan HJ, Lamont RE, Majewski J, Parboosingh JS, Prasad AN, Rupar CA, Schwartzentruber J, Smith AC, Tétreault M, FORGE Canada Consortium, Care4Rare Canada Consortium, Innes AM, Boycott KM (2017) Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin Genet 92:281–289
Baux D, Blanchet C, Hamel C, Meunier I, Larrieu L, Faugère V, Vaché C, Castorina P, Puech B, Bonneau D, Malcolm S, Claustres M, Roux AF (2014) Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum Mutat 35:1179–1186
Baux D, Vaché C, Blanchet C, Willems M, Baudoin C, Moclyn M, Faugère V, Touraine R, Isidor B, Dupin-Deguine D, Nizon M, Vincent M, Mercier S, Calais C, García-García G, Azher Z, Lambert L, Perdomo-Trujillo Y, Giuliano F, Claustres M, Koenig M, Mondain M, Roux AF (2017) Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci Rep 7:16783
Beesley CE, Meaney CA, Greenland G, Adams V, Vellodi A, Young EP, Winchester BG (2001) Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet 109:503–511
Beutler E, Westwood B, Prchal JT, Vaca G, Bartsocas CS, Baronciani L (1992) New glucose-6-phosphate dehydrogenase mutations from various ethnic groups. Blood 80:255–256
Bharadwaj AK, Kasztejna JP, Huq S, Berson EL, Dryja TP (2000) Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp Eye Res 71:173–181
Bonnet C, Grati M, Marlin S, Levilliers J, Hardelin JP, Parodi M, Niasme-Grare M, Zelenika D, Délépine M, Feldmann D, Jonard L, El-Amraoui A, Weil D, Delobel B, Vincent C, Dollfus H, Eliot MM, David A, Calais C, Vigneron J, Montaut-Verient B, Bonneau D, Dubin J, Thauvin C, Duvillard A, Francannet C, Mom T, Lacombe D, Duriez F, Drouin-Garraud V, Thuillier-Obstoy MF, Sigaudy S, Frances AM, Collignon P, Challe G, Couderc R, Lathrop M, Sahel JA, Weissenbach J, Petit C, Denoyelle F (2011) Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis 6:21
Bonnet C, Riahi Z, Chantot-Bastaraud S, Smagghe L, Letexier M, Marcaillou C, Lefèvre GM, Hardelin JP, El-Amraoui A, Singh-Estivalet A, Mohand-Saïd S, Kohl S, Kurtenbach A, Sliesoraityte I, Zobor D, Gherbi S, Testa F, Simonelli F, Banfi S, Fakin A, Glavač D, Jarc-Vidmar M, Zupan A, Battelino S, Martorell Sampol L, Claveria MA, Catala Mora J, Dad S, Møller LB, Rodriguez Jorge J, Hawlina M, Auricchio A, Sahel JA, Marlin S, Zrenner E, Audo I, Petit C (2016) An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur J Hum Genet 24:1730–1738
Booth KT, Kahrizi K, Babanejad M, Daghagh H, Bademci G, Arzhangi S, Zareabdollahi D, Duman D, El-Amraoui A, Tekin M, Najmabadi H, Azaiez H, Smith RJ (2018) Variants in CIB2 cause DFNB48 and not USH1J. Clin Genet 93:812–821
Boskabadi H, Zakerihamidi M, Moradi A, Mehdi Bakhshaee M (2018) Risk factors for sensorineural hearing loss in neonatal hyperbilirubinemia. Iran J Otorhinolaryngol 30:195–202
Bunge S, Wedemann H, David D, Terwilliger DJ, van den Born LI, Aulehla-Scholz C, Samanns C, Horn M, Ott J, Schwinger E, Schinzel A, Denton MJ, Gal A (1993) Molecular analysis and genetic map** of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics 17:230–233
Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ, Salvetti AP, Fischer MD, Aylward JW, Barnard AR, Jolly JK, Luo E, Lujan BJ, Ong T, Girach A, Black GCM, Gregori NZ, Davis JL, Rosa PR, Lotery AJ, Lam BL, Stanga PE, MacLaren RE (2020) Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med 26:354–359
Chassaing N, Martin L, Mazereeuw J, Barrié L, Nizard S, Bonafé JL, Calvas P, Hovnanian A (2004) Novel ABCC6 mutations in pseudoxanthoma elasticum. J Invest Dermatol 122:608–613
Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H (2013) Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 132:1077–1130
Cremers FP, Kimberling WJ, Külm M, de Brouwer AP, van Wijk E, te Brinke H, Cremers CW, Hoefsloot LH, Banfi S, Simonelli F, Fleischhauer JC, Berger W, Kelley PM, Haralambous E, Bitner-Glindzicz M, Webster AR, Saihan Z, de Baere E, Leroy BP, Silvestri G, McKay GJ, Koenekoop RK, Millan JM, Rosenberg T, Joensuu T, Sankila EM, Weil D, Weston MD, Wissinger B, Kremer H (2007) Development of a genoty** microarray for Usher syndrome. J Med Genet 44:153–160
Dammeyer J (2014) Deafblindness: a review of the literature. Scand J Public Health 42:554–562
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C (2009) Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37:e67
Diñeiro M, Capín R, Cifuentes GÁ, Fernández-Vega B, Villota E, Otero A, Santiago A, Pruneda PC, Castillo D, Viejo-Díaz M, Hernando I, Durán NS, Álvarez R, Lago CG, Ordóñez GR, Fernández-Vega Á, Cabanillas R, Cadiñanos J (2020) Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options. Acta Ophthalmol 98:1034–1048
DiStefano MT, Hemphill SE, Oza AM, Siegert RK, Grant AR, Hughes MY, Cushman BJ, Azaiez H, Booth KT, Chapin A, Duzkale H, Matsunaga T, Shen J, Zhang W, Kenna M, Schimmenti LA, Tekin M, Rehm HL, Tayoun ANA, Amr SS, ClinGen Hearing Loss Clinical Domain Working Group (2019) ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet Med 21:2239–2247
Downie L, Halliday J, Burt R, Lunke S, Lynch E, Martyn M, Poulakis Z, Gaff C, Sung V, Wake M, Hunter MF, Saunders K, Rose E, Lewis S, Jarmolowicz A, Phelan D, Rehm HL, Alliance MGH, Amor D (2020) Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur J Hum Genet 28:587–596
Duzkale H, Shen J, McLaughlin H, Alfares A, Kelly MA, Pugh TJ, Funke BH, Rehm HL, Lebo MS (2013) A systematic approach to assessing the clinical significance of genetic variants. Clin Genet 84:453–463
Ebermann I, Wiesen MH, Zrenner E, Lopez I, Pigeon R, Kohl S, Löwenheim H, Koenekoop RK, Bolz HJ (2009) GPR98 mutations cause Usher syndrome type 2 in males. J Med Genet 46:277–280
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J (1998) Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280:1753–1757
Gao FJ, Wang DD, Cheng F, Sun HX, Hu FY, Xu P, Li J, Liu W, Qi YH, Li W, Wang M, Zhang S, Xu GZ, Chang Q, Wu JH (2021) Prevalence and genetic–phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br J Ophthalmol 105:87–92
Géléoc GGS, El-Amraoui A (2020) Disease mechanisms and gene therapy for Usher syndrome. Hear Res 394:107932
Hart MR, Biesecker BB, Blout CL, Christensen KD, Amendola LM, Bergstrom KL, Biswas S, Bowling KM, Brothers KB, Conlin LK, Cooper GM, Dulik MC, East KM, Everett JN, Finnila CR, Ghazani AA, Gilmore MJ, Goddard KAB, Jarvik GP, Johnston JJ, Kauffman TL, Kelley WV, Krier JB, Lewis KL, McGuire AL, McMullen C, Ou J, Plon SE, Rehm HL, Richards CS, Romasko EJ, Miren Sagardia A, Spinner NB, Thompson ML, Turbitt E, Vassy JL, Wilfond BS, Veenstra DL, Berg JS, Green RC, Biesecker LG, Hindorff LA (2019) Secondary findings from clinical genomic sequencing: prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet Med 21:1100–1110
Hombach D, Schuelke M, Knierim E, Ehmke N, Schwarz JM, Fischer-Zirnsak B, Seelow D (2019) MutationDistiller: user-driven identification of pathogenic DNA variants. Nucleic Acids Res 47:W114–W120
Jaijo T, Aller E, Oltra S, Beneyto M, Nájera C, Ayuso C, Baiget M, Carballo M, Antiñolo G, Valverde D, Moreno F, Vilela C, Perez-Garrigues H, Navea A, Millán JM (2006) Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum Mutat 27:290–291
Jiang L, Liang X, Li Y, Wang J, Zaneveld JE, Wang H, Xu S, Wang K, Wang B, Chen R, Sui R (2015) Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J Rare Dis 10:110
Jouret G, Poirsier C, Spodenkiewicz M, Jaquin C, Gouy E, Arndt C, Labrousse M, Gaillard D, Doco-Fenzy M, Lebre AS (2019) Genetics of Usher syndrome: new insights from a meta-analysis. Otol Neurotol 40:121–129
Kajiwara K, Berson EL, Dryja TP (1994) Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 264:1604–1608
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT (2017) Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 19:249–255
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443
Khalaileh A, Abu-Diab A, Ben-Yosef T, Raas-Rothschild A, Lerer I, Alswaiti Y, Chowers I, Banin E, Sharon D, Khateb S (2018) The genetics of Usher syndrome in the Israeli and Palestinian populations. Invest Ophthalmol vis Sci 59:1095–1104
Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K, Cohn ES, Weleber RG, Stone EM, Smith RJ (2010) Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet Med 12:512–516
Kooshavar D, Razipour M, Movasat M, Keramatipour M (2018) Targeted next generation sequencing identified a novel mutation in MYO7A causing Usher syndrome type 1 in an Iranian consanguineous pedigree. Int J Pediatr Otorhinolaryngol 104:10–13
Le Quesne SP, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, Emmerson J, Haralambous E, Hughes Y, Steel KP, Luxon LM, Webster AR, Bitner-Glindzicz M (2012) Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet 49:27–36
Lentz JJ, Pan B, Ponnath A, Tran CM, Nist-Lund C, Galvin A, Goldberg H, Robillard KN, Jodelka FM, Farris HE, Huang J, Chen T, Zhu H, Zhou W, Rigo F, Hastings ML, Géléoc GSG (2020) Direct delivery of antisense oligonucleotides to the middle and inner ear improves hearing and balance in Usher mice. Mol Ther 28:2662–2676
Lewis TR, Shores CR, Cady MA, Hao Y, Arshavsky VY, Burns ME (2020) The F220C and F45L rhodopsin mutations identified in retinitis pigmentosa patients do not cause pathology in mice. Sci Rep 10:7538
Liquori A, Vaché C, Baux D, Blanchet C, Hamel C, Malcolm S, Koenig M, Claustres M, Roux AF (2016) Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum Mutat 37:184–193
Liu L, Li H, Shi L (2017) Alström syndrome with novel ALMS mutations: a case report. Exp Clin Endocrinol Diabetes Rep 5:e10-13
Mallory DP, Gutierrez E, Pinkevitch M, Klinginsmith C, Comar WD, Roushar FJ, Schlebach JP, Smith AW, Jastrzebska B (2018) The retinitis pigmentosa-linked mutations in transmembrane helix 5 of rhodopsin disrupt cellular trafficking regardless of oligomerization state. Biochemistry 57:5188–5201
Mazzoli M, van Camp G, Newton V, Giarbini N, Declau F, Parving A (2003) Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol Med 1:148–150
Myers KA, Nasioulas S, Boys A, McMahon JM, Slater H, Lockhart P, Sart DD, Scheffer IE (2018) ADGRV1 is implicated in myoclonic epilepsy. Epilepsia 59:381–388
Nadeau JH (2001) Modifier genes in humans and mice. Nat Rev Genet 2:165–174
Nagel-Wolfrum K, Baasov T, Wolfrum U (2014) Therapy strategies for Usher syndrome type 1C in the retina. Adv Exp Med Biol 801:741–747
Namburi P, Ratnapriya R, Khateb S, Lazar CH, Kinarty Y, Obolensky A, Erdinest I, Marks-Ohana D, Pras E, Ben-Yosef T, Newman H, Gross M, Swaroop A, Banin E, Sharon D (2016) Bi-allelic truncating mutations in CEP78, encoding centrosomal protein 78, cause cone-rod degeneration with sensorineural hearing loss. Am J Hum Genet 99:777–784
Neuhaus C, Eisenberger T, Decker C, Nagl S, Blank C, Pfister M, Kennerknecht I, Müller-Hofstede C, Charbel Issa P, Heller R, Beck B, Rüther K, Mitter D, Rohrschneider K, Steinhauer U, Korbmacher HM, Huhle D, Elsayed SM, Taha HM, Baig SM, Stöhr H, Preising M, Markus S, Moeller F, Lorenz B, Nagel-Wolfrum K, Khan AO, Bolz HJ (2017) Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: copy number variations, phenocopies, a predominant target for translational read-through, and PEX26 mutated in Heimler syndrome. Mol Genet Genomic Med 5:531–552
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874
Oishi M, Oishi A, Gotoh N, Ogino K, Higasa K, Iida K, Makiyama Y, Morooka S, Matsuda F, Yoshimura N (2014) Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest Ophthalmol vis Sci 55:7369–7735
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Murry JB, Hasadsri L, Nara K, Kenna M, Booth KT, Azaiez H, Griffith A, Avraham KB, Kremer H, Rehm HL, Amr SS, Abou Tayoun AN, ClinGen Hearing Loss Clinical Domain Working Group (2018) Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat 39:1593–1613
Pertea M, Lin X, Salzberg SL (2001) GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 29:1185–1190
Peter VG, Quinodoz M, Sadio S, Held S, Rodrigues M, Soares M, Sousa AB, Coutinho Santos L, Damme M, Rivolta C (2021) New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum Mutat 42:261–271
Pierrache LH, Hartel BP, van Wijk E, Meester-Smoor MA, Cremers FP, de Baere E, de Zaeytijd J, van Schooneveld MJ, Cremers CW, Dagnelie G, Hoyng CB, Bergen AA, Leroy BP, Pennings RJ, van den Born LI, Klaver CC (2016) Visual prognosis in USH2A-associated retinitis pigmentosa is worse for patients with Usher syndrome type IIa than for those with nonsyndromic retinitis pigmentosa. Opthalmology 123:1151–1160
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, **ao R, Ding Y, **a F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE, Yang Y, Lupski JR (2017) Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 376:21–31
Puffenberger EG, **ks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, Mahoney MH, Miller CJ, Nair DT, Politi KA, Worcester KN, Setton RA, Dipiazza R, Sherman EA, Eastman JT, Francklyn C, Robey-Bond S, Rider NL, Gabriel S, Morton DH, Strauss KA (2012) Genetic map** and exome sequencing identify variants associated with five novel diseases. PLoS ONE 7:e28936
Ratnapriya R, Zhan X, Fariss RN, Branham KE, Zipprer D, Chakarova CF, Sergeev YV, Campos MM, Othman M, Friedman JS, Maminishkis A, Waseem NH, Brooks M, Rajasimha HK, Edwards AO, Lotery A, Klein BE, Truitt BJ, Li B, Schaumberg DA, Morgan DJ, Morrison MA, Souied E, Tsironi EE, Grassmann F, Fishman GA, Silvestri G, Scholl HP, Kim IK, Ramke J, Tuo J, Merriam JE, Merriam JC, Park KH, Olson LM, Farrer LA, Johnson MP, Peachey NS, Lathrop M, Baron RV, Igo RP Jr, Klein R, Hagstrom SA, Kamatani Y, Martin TM, Jiang Y, Conley Y, Sahel JA, Zack DJ, Chan CC, Pericak-Vance MA, Jacobson SG, Gorin MB, Klein ML, Allikmets R, Iyengar SK, Weber BH, Haines JL, Léveillard T, Deangelis MM, Stambolian D, Weeks DE, Bhattacharya SS, Chew EY, Heckenlively JR, Abecasis GR, Swaroop A (2014) Rare and common variants in extracellular matrix gene fibrillin 2 (FBN2) are associated with macular degeneration. Hum Mol Genet 23:5827–5837
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323
Rethanavelu K, Fung JLF, Chau JFT, Pei SLC, Chung CCY, Mak CCY, Luk HM, Chung BHY (2020) Phenotypic and mutational spectrum of 21 Chinese patients with Alström syndrome. Am J Med Genet A 182:279–288
Riazuddin S, Nazli S, Ahmed ZM, Yang Y, Zulfiqar F, Shaikh RS, Zafar AU, Khan SN, Sabar F, Javid FT, Wilcox ER, Tsilou E, Boger ET, Sellers JR, Belyantseva IA, Riazuddin S, Friedman TB (2008) Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat 29:502–511
Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, Yousaf R, Sinha GP, Lee S, Terrell D, Hegde RS, Ali RA, Anwar S, Andrade-Elizondo PB, Sirmaci A, Parise LV, Basit S, Wali A, Ayub M, Ansar M, Ahmad W, Khan SN, Akram J, Tekin M, Riazuddin S, Cook T, Buschbeck EK, Frolenkov GI, Leal SM, Friedman TB, Ahmed ZM (2012) Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 44:1265–1271
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Rio Frio T, McGee TL, Wade NM, Iseli C, Beckmann JS, Berson EL, Rivolta C (2009) A single-base substitution within an intronic repetitive element causes dominant retinitis pigmentosa with reduced penetrance. Hum Mutat 30:1340–1347
Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP (2006) Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat 27:644–653
Rose AM, Shah AZ, Venturini G, Krishna A, Chakravarti A, Rivolta C, Bhattacharya SS (2016) Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci Rep 6:19450
Roux AF, Faugère V, Le Guédard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M, Collaboration FUS (2006) Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J Med Genet 43:763–768
Sabbaghi H, Daftarian N, Suri F, Mirrahimi M, Madani S, Sheikhtaheri A, Khorrami F, Saviz P, Zarei Nejad M, Tivay A, Shahriari HA, Maleki A, Ahmadi SS, Sargazi M, Cremers FPM, Najafi M, Vona B, Haaf T, Bahena-Carbajal P, Moghadasi A, Naraghi H, Yaseri M, Kheiri B, Kalantarion M, Sabbaghi E, Salami M, Pazooki L, Zendedel K, Mojarrab S, Ahmadieh H (2020) The first inherited retinal disease registry in Iran: research protocol and results of a pilot study. Arch Iran Med 23:445–454
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362
Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, Gabriel SB, Belkadi A, Boisson B, Abel L, Clark AG, Greater Middle East Variome Consortium, Alkuraya FS, Casanova JL, Gleeson JG (2016) Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet 48:1071–1076
Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174
Simonelli F, Testa F, de Crecchio G, Rinaldi E, Hutchinson A, Atkinson A, Dean M, D’Urso M, Allikmets R (2000) New ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Invest Ophthalmol vis Sci 41:892–897
Sliesoraityte I, Peto T, Mohand-Said S, Sahel JA (2015) Novel grading system for quantification of cystic macular lesions in Usher syndrome. Orphanet J Rare Dis 10:157
Sloan-Heggen CM, Babanejad M, Beheshtian M, Simpson AC, Booth KT, Ardalani F, Frees KL, Mohseni M, Mozafari R, Mehrjoo Z, Jamali L, Vaziri S, Akhtarkhavari T, Bazazzadegan N, Nikzat N, Arzhangi S, Sabbagh F, Otukesh H, Seifati SM, Khodaei H, Taghdiri M, Meyer NC, Daneshi A, Farhadi M, Kahrizi K, Smith RJ, Azaiez H, Najmabadi H (2015) Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet 52:823–829
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJH (2016) Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 135:441–450
Soens ZT, Branch J, Wu S, Yuan Z, Li Y, Li H, Wang K, Xu M, Rajan L, Motta FL, Simões RT, Lopez-Solache I, Ajlan R, Birch DG, Zhao P, Porto FB, Sallum J, Koenekoop RK, Sui R, Chen R (2017) Leveraging splice-affecting variant predictors and a minigene validation system to identify Mendelian disease-causing variants among exon-captured variants of uncertain significance. Hum Mutat 38:1521–1533
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN (2014) The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133:1–9
Stiff HA, Sloan-Heggen CM, Ko A, Pfeifer WL, Kolbe DL, Nishimura CJ, Frees KL, Booth KT, Wang D, Weaver AE, Azaiez H, Kamholz J, Smith RJH, Drack AV (2020) Is it Usher syndrome? Collaborative diagnosis and molecular genetics of patients with visual impairment and hearing loss. Ophthalmic Genet 41:151–158
Tasliyurt T, Yigit S, Rustemoglu A, Gul U, Ates O (2013) Common MEFV gene mutations in Turkish patients with Behcet’s disease. Gene 530:100–103
Tompson SW, Young TL (2017) Assaying the effects of splice site variants by exon trap** in a mammalian cell line. Bio Protoc 7:e2281
Vaché C, Besnard T, le Berre P, García-García G, Baux D, Larrieu L, Abadie C, Blanchet C, Bolz HJ, Millan J, Hamel C, Malcolm S, Claustres M, Roux AF (2012) Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum Mutat 33:104–108
Watanabe S, Umeki N, Ikebe R, Ikebe M (2008) Impacts of Usher syndrome type IB mutations on human myosin VIIa motor function. Biochemistry 47:9505–9513
Yang Y, Muzny DM, **a F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312:1870–1879
Yeo G, Burge CB (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11:377–394
Acknowledgements
We appreciate the collaboration of the probands and their families in this study. This study was performed with the help of "The Iranian National Registry of Inherited Retinal Dystrophy (IRDReg)" (code number of IR.SBMU.ORC.REC.1396.15) and supported by the Deputy of Research and Technology at Shahid Beheshti University of Medical Sciences, Tehran, Iran (http://dregistry.sbmu.ac.ir).
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by Intramural Funding (fortüne) at the University of Tübingen (2545-1-0 to BV) and the Ministry of Science, Research and Art Baden-Württemberg (to BV).
Author information
Authors and Affiliations
Contributions
PB, FS, BV, and TH designed the study. ND, RM, MM, HS, HA, HB, CVM, VCG, HD, EA, MAS, and FS recruited probands and provided clinical information. PB, PL, DV, AR, JD, MH, AK, TR, and SH performed the experiments. PB, SK, RZO, LK, FS, and BV analyzed the data. PB, BV, and TH wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This study was approved by the ethical review boards at the Medical Faculty of Würzburg University, Germany (approval number 46/15), the National Institute of Rehabilitation Luis Guillermo Ibarra (INR), Mexico (no. 12/13), and the Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Consent to participate
Consent for participation was received from the probands and their families.
Consent for publication
Consent for publication was received from the probands and their families.
Animal experiments
Not applicable as no animal experiments were performed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bahena, P., Daftarian, N., Maroofian, R. et al. Unraveling the genetic complexities of combined retinal dystrophy and hearing impairment. Hum Genet 141, 785–803 (2022). https://doi.org/10.1007/s00439-021-02303-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02303-1