
Abstract
The extensive clinical and genetic heterogeneity of congenital limb malformation calls for comprehensive genome-wide analysis of genetic variation. Genome sequencing (GS) has the potential to identify all genetic variants. Here we aim to determine the diagnostic potential of GS as a comprehensive one-test-for-all strategy in a cohort of undiagnosed patients with congenital limb malformations. We collected 69 cases (64 trios, 1 duo, 5 singletons) with congenital limb malformations with no molecular diagnosis after standard clinical genetic testing and performed genome sequencing. We also developed a framework to identify potential noncoding pathogenic variants. We identified likely pathogenic/disease-associated variants in 12 cases (17.4%) including four in known disease genes, and one repeat expansion in HOXD13. In three unrelated cases with ectrodactyly, we identified likely pathogenic variants in UBA2, establishing it as a novel disease gene. In addition, we found two complex structural variants (3%). We also identified likely causative variants in three novel high confidence candidate genes. We were not able to identify any noncoding variants. GS is a powerful strategy to identify all types of genomic variants associated with congenital limb malformation, including repeat expansions and complex structural variants missed by standard diagnostic approaches. In this cohort, no causative noncoding SNVs could be identified.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The repertoire of diagnostic tests in human genetics is as diverse as the types of genetic alterations they were developed to detect (Berisha et al. 2020). Through the development of Next Generation Sequencing technologies (NGS) sequencing has become several orders of magnitude faster and cheaper. This has led to an enormous increase in the efficiency of genetic testing (Levy and Myers 2016). NGS quickly found its way from research applications to the clinic: today, panel and exome sequencing are elements of the routine diagnostics in genetic medicine (Deciphering Developmental Disorders Study 2017). Despite these significant advances, classical genetic testing methods such as chromosomal microarray analysis (CMA) and Sanger sequencing remain part of the standard diagnostic arsenal. This is because NGS-based gene panels often do not detect structural variants such as inversions and translocations, or fail to determine repeat lengths (Berisha et al. 2020). The goal of detecting all types of genetic variation in a single test can theoretically be achieved by short-read based genome sequencing (GS) (Xue et al. 2015). While there are some very encouraging proof of concept studies for the use of GS in individuals with intellectual disability (Lindstrand et al. 2019), GS is not yet part of the clinical routine and there is a lack of systematic studies on the benefits of such tests for individuals with congenital malformations.
A major limitation of panel and exome sequencing approaches is that they usually do not cover 98% of the genome which is noncoding, and are, hence, unable to detect deep intronic splice variants or intergenic regulatory variants. Therefore, over 40% of individuals with genetic diseases receive no molecular diagnosis after standard testing (Gilissen et al. 2014). This is likely because the noncoding sequence has largely been ignored despite most nucleotides and single nucleotide variants being noncoding. The two main challenges that currently hamper the medical interpretation of noncoding variants are the poor understanding of the “regulatory code” of the noncoding genome and the large number of noncoding variants in each individual that renders classical functional work-up strategies impossible.
In this study, we aimed to determine the diagnostic potential of GS as a comprehensive one-test-for-all strategy in a cohort of 69 unsolved patients with congenital limb malformations. We also attempted to develop a framework to prioritize the large number of noncoding variants identified in the GS studies by combining mouse genetic and human functional epigenetic data with in vivo-validated enhancer sequences.
Materials and methods
Study design
Patients affected with malformations of two limbs, or two individuals from a family, each affected with a malformation of at least one limb were recruited (Supplementary Fig. 1). Exclusion criteria included a molecularly established genetic diagnosis, a suspected diagnosis of amniotic band syndrome, or an isolated fifth finger clinodactyly. A convenience set of samples was collected from the patients of the Department of Hand Surgery of the Katholisches Kinderkrankenhaus Wilhelmstift Hamburg and the Institute of Medical Genetics and Human Genetics of the Charité (IMG)—Universitätsmedizin Berlin. This sample-set was compiled with cases that were sent to the IMG by external physicians for diagnostic purposes. The sample-set was fixed before conducting GS.
Included patients
We included 69 patients in this study (Supplementary Table 1). We sequenced the index case and both parents in 64 cases, the index and one parent in one case, and only the index in four cases (parental DNA was not available for testing). In one case, we additionally sequenced a sibling. In five cases, one parent showed a limb malformation comparable to the index. In one case featuring ectrodactyly and apparently unaffected parents and grandparents, a maternal grand-uncle was affected, who was also sequenced. In 60 cases no family member other than the index was reported to show a limb malformation.
Phenoty** and conventional genetic testing
Limb malformations were phenotyped based on photographs and radiographs by a panel of medical professionals including expert clinical geneticists. Phenotypes were described as per Human Phenotype Ontology (HPO) terminology.
Based on a patient's phenotype, genes were selected for sequencing by medical geneticists. Sample preparation and Sanger-sequencing were performed using standard procedures. High resolution (1 M oligo) CMA was performed as described previously (Flöttmann et al. 2018).
Genome sequencing and variant calling
Paired-end PCR-free GS was performed by Macrogen Inc. (South-Korea) using a HiSeq X Ten platform. DNA preparation, sequencing, and sequence data processing were performed according to Macrogen’s standard protocol (coverage 30× and read length 150 bp).
The FASTQ files were transferred to the Core Unit Bioinformatics of the Berlin Institute of Health (CUBI) for variant calling. Files were further processed and securely stored in the System for Omics Data Analysis and Retrieval (SODAR) (Nieminen et al. 2020). GATK HC was used to call simple nucleotide variants, while structural variants were called using Delly2, PopDel, and ERDS/SV2. Afterwards, variants were processed and annotated by the VarFish platform (Holtgrewe et al. 2020). Variants were mapped according to the hg19 reference genome.
Variant filtration
Coding SNVs and SVs
Each index case was filtered as a singleton, regardless of the availability of family data. If parental samples were available, a trio-based filtration approach was additionally performed. Male-index trio-cases were also filtered for hemizygous X-chromosomal variants.
Simple nucleotide variant filtration was performed on the VarFish platform (Version v0.17.2) (Holtgrewe et al. 2020). We filtered GS data for non-synonymous exonic and splice variants using default settings for read depth, allelic balance, and read quality. Allele counts were set as described in Supplementary Table 2. SNVs that passed filtration were exported as variant calling files (VCF). For evaluation of variant pathogenicity, VCFs were uploaded to MutationDistiller and Exomiser. The first ten results were exported for semi-automated, in-depth analysis (see Supplementary Fig. 2 for details). We also tested for truncating or probable LoF (CADD > 20) variants affecting the same gene with a pLI > 0.9 in at least two independent patients.
Structural variant filtration was performed as described in Supplementary Table 3. The minimal size for structural variant filtration was 1500 bp. Whenever we obtained more than 30 structural variants after initial filtering, we increased the number of minimally covered informative reads from 2 to 5. Each SV passing filtration was judged manually with the information provided by the IGV-Browser and UCSC (see Supplementary Fig. 3 for details).
All findings were evaluated at weekly clinical meetings.
Analysis of noncoding variants: limb regulome
We defined a limb-specific potential regulome to filter and interpret non-coding variants. For this purpose, we created a list of 1719 genes involved in embryonic limb development based on data from the Mouse Genome Informatics (MGI) database and entries in OMIM. We defined the human limb regulome as the following: 1. all conserved (phyloP > 1,3) variants, 2. those located within the same topologically associating domain (TAD) (as determined in human fibroblasts (Dixon et al. 2012)) as a limb gene, 3. those that were marked by an H3K27 acetylation peak in human limb buds (Cotney et al. 2013). We also included the validated enhancer elements of the VISTA Enhancer Browser. Coding and noncoding SNVs were filtered for rare variants (frequency < 0.1%) and were considered to be potentially affecting the same regulatory element if they were either less than 300 bp apart or positioned in the same established enhancer.
Whenever we identified rare, potentially pathogenic heterozygous coding variants of genes associated with a recessive limb phenotype in individuals featuring at least a partial overlap with that phenotype, we also screened for in trans conserved non-coding variants with a MAF < 3% affecting the same TAD.
Results
We collected a cohort of 69 individuals affected with congenital limb malformations. All individuals had previously gone through our clinical genetics routine pipeline including clinical examination, candidate gene testing, and CMA. We then performed GS as a comprehensive one-test-for-all strategy.
In total, we identified 333,163,643 single nucleotide variants (SNVs) among the 69 sequenced index patients, of which 7,020,766 were either coding or flanking coding elements by 10 bp or less. 326,142,877 were noncoding SNVs, of which 19,362 were rare (gnomAD AF < 0.01). 21,369 of the coding SNV calls were classified by Jannovar to be of at least moderate relevance (missense and truncating), and 1429 to be of high relevance (truncating only). VarFish filtering returned 5761 SNVs. Filtering by Exomizer and MutationTaster identified 433 potentially pathogenic coding calls among these, of which 174 were high-quality calls suitable for further evaluation.
30,062, SNVs resulted in the potential loss-of-function of probably haploinsufficient genes. 49 of these affected the same gene in two unrelated index individuals (Supplementary Fig. 2).
We also analyzed the structural variants in 68 of the 69 index patients. 55 cases were filtered as trios with unaffected parents and moderate filter settings. Five were filtered as trios with another affected relative and moderate filter settings. Stricter filter settings were chosen for 9 trios because moderate filter settings produced an unmanageable amount of SV calls. 3 cases were analyzed as singletons. Individuals I1, I2, I3 did not yield any results, I4 was excluded from the SV analysis because too many SVs were called even with stricter filter settings due to poor data quality.
Of the 1,555,426 SVs, 633 SVs passed the filtering by VarFish, of which 222 were inversions, 288 deletions, 76 duplications, and 47 breakpoints of potential translocations.
417 of these SVs were excluded because they were of poor calling quality or because they were inherited from an unaffected parent. We then manually inspected the remaining 216 SVs. Segregation analysis in the parents was performed by qPCR after comparing candidate CNVs with known limb genes according to the Human Phenotype Ontology, cross-species phenotype comparison, mouse models, gene expression data (Cao et al. 2019), limb enhancer elements (Visel et al. 2009), and the local topological associating domain (TAD) architecture of the locus (Dixon et al. 2012; Cao et al. 2019). As a result, we identified 30 promising variants (Supplementary Fig. 3).
Variants in four known, limb malformation associated genes
We identified pathogenic variants in established disease genes in four individuals (I5, I6, I7, I8, Supplementary Fig. 4), which we confirmed by Sanger sequencing. These included a missense variant in FGFR1, already described in the literature (Muenke et al. 2014), and three previously undescribed variants in the genes FGFR2, GLI3, and BHLHA9. In all four cases, we classified the variants as (likely) pathogenic according to the criteria of the American College of Medical Genetics and Genomics (ACMG), based on the type of variant and the phenotype of the patient. All variants were inherited (note that the mother of I8, was not radiographically phenotyped, which is necessary to diagnose mild FGFR2-associated phenotypes (Flöttmann et al. 2015)).
Repeat expansions of HOXD13
In individual I9, featuring brachy-poly-syndactyly, we detected a repeat expansion by eight alanines on the HOXD13 allele inherited from his affected mother, already described as pathogenic in the literature (Brison et al. 2014), and a polymorphic repeat expansion by only one alanine on the paternal HOXD13 allele (Supplementary Fig. 5). These findings were confirmed by conventional HOXD13 microsatellite analysis.
Structural variants at known disease loci
In individual I10 with bilateral upper and lower limb ectrodactyly, we identified an inversion of 105 kb (chr10: 103,321,526–103,426,609) flanked by two deletions (chr10:103,319,219–103,321,525 and chr10:103,426,610–103,436,718) at the split-hand foot malformation locus 3 (SHFM3) on chr10q24 inherited from his unaffected mother (Fig. 1, Supplementary Fig. 6). His affected great-uncle also carried the inversion. The variant overlaps with the most common duplications associated with ectrodactyly (de Mollerat et al. 2003; Klopocki et al. 2012). The minimal overlap** region of pathogenic SHFM3 duplications includes BTRC, POLL, and DPCD (Holder-Espinasse et al. 2019). The inversion described here is copy number neutral, suggesting that positional effects rather than gene dosage might be responsible for the phenotype. It includes a topologically associating domain boundary (Holder-Espinasse et al. 2019) and is likely to change the enhancer landscape at the SHFM3 locus leading to FGF8 misregulation causing ectrodactyly.

Inversion-deletion at SHFM3 locus: a pedigree, N.T. not tested. b feet of grand-uncle (II-3). c hands and feet of the index patient (IV-1). d genomic architecture of SHFM3. e GS data of the family, note the presence of an inversion (chr10: 103,321,526–103,426,609) flanked by deletions (chr10:103,319,219–103,321,525 and chr10:103,426,610–103,436,718) on either site
In individual I11, featuring bilateral mirror-image polydactyly of the hands and feet (Fig. 2a), CMA had detected a 300 kb amplification on chr7q36.1. We initially classified the variant as a variant of unknown significance, because the individual’s phenotype did not match that of an individual with muscular hypertrophy, reported to have a similarly sized and positioned duplication (Kroeldrup et al. 2012).

a Pedigree and phenotype of individual I11. b Potential neo-TAD at the fusion site. c Breakpoint and fusion sites between regions from chr7 and chr9
The amplification was identified again using GS. However, sequencing revealed that it was part of a complex structural variant containing two overlap** duplications (dup1 and dup2) at chr7q36.1 (Supplementary Fig. 7). The smaller dup2 shares the central breakpoint with dup1. The distal breakpoint of dup2 is positioned within dup1 in intron 1 of SHH, at chr7:155,603,964. GS data showed that the duplicons were not positioned in tandem, but are both fused to sequences originating from chr9p24.1. The distal breakpoint of dup2 was fused to intron 5 of GLDC and the distal breakpoint of dup1 to intron 8 of KDM4C (Fig. 2c; Supplementary Fig. 7). Analysis of the parents by Sanger sequencing showed that the structural variant occurred de novo in individual I11.
The mirror-image polydactyly of individual I11 shows striking phenotypic overlap with Laurin–Sandrow syndrome, which is caused by duplications of the SHH regulator ZRS, positioned in intron 5 of LMBR1 on chr7p36.3, resulting in ectopic expression of SHH in the embryonic limb (Lohan et al. 2014). Both duplications do not include the ZRS and duplications of SHH itself have not been described to cause Laurin–Sandrow syndrome. However, a duplicated fragment containing SHH that is inserted into another domain, as observed in the de novo SV of I11, makes an ectopic expression of SHH in the embryonic limb very likely. We assume that the formation of an SHH-KDM4C neo-TAD, resulting in the misregulation of SHH by KDM4C-enhancers in the limb mesenchyme (note the known expression of KDM4C in embryonic vertebrate limb buds) is the most likely explanation for such an ectopic SHH-expression (Fig. 2b).
Therefore, we re-classified the complex SV involving SHH in individual I11 with bilateral mirror-image polydactyly as causative.
Establishing UBA2 as a novel disease gene
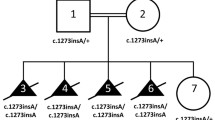
We also identified variants in new candidate genes. Two unrelated individuals with isolated split hand malformation featured different heterozygous frameshift variants in the ubiquitin-like modifier-activating enzyme 2 (UBA2) (Fig. 3a, b). Individual I12 harboured the de novo variant NM_005499.3(UBA2):c.1355_1356delTG;p.(Val452Alafs*6). Individual I13 inherited the variant NM_005499.3(UBA2):c.34_37delGCTG;p.(Ala12Argfs*34) from his apparently unaffected mother (no radiographs of her hands were available).

UBA2 variants and ectrodactyly. a–c Patients with likely pathogenic UBA2 variants upper panels: pedigrees, N.T.: not tested; middle panels: characteristic limb malformations, lower panels: sequencing data. d conservation of Asp50 mutated in individual I14, numbers indicate amino acid residues, yellow bars highlight positions tested by Olsen et al. to cause loss of function when substituted by alanine (Olsen et al. 2010)
We classified the pathogenicity of these variants according to the ACMG guidelines. Both are null variants of UBA2 which has a pLI-score of 1 (PVS1). c.1355_1356delTG occurred de novo in an individual with a negative family history (PS2). The variants are absent from the 1000 Genomes Project and the Exome Aggregation Consortium databases (PM2) and were predicted to be pathogenic by MutationTaster (PP3). UBA2 variants have recently been described in individuals with ectrodactyly (Chowdhury et al. 2014; Abe et al. 2018; Yamoto et al. 2019; Aerden et al. 2020). Hence, we regarded UBA2 as a disease-associated gene and these variants as pathogenic (1PVS(+ 1PS) + 1PM + 1PP).
These findings prompted subsequent Sanger sequencing of UBA2 in 24 unrelated families with ectrodactyly, who have been tested negative for variants in the established SHFM loci/genes. In one individual (I14) with unilateral split-hand malformation (Fig. 3c), we identified the missense variant NM_005499.3(UBA2):c.149A > G;p (Asp50Gly). The daughter of I14 and her son also feature ectrodactyly (PP4), but were unavailable for testing. Asp50 is part of a consecutive 15 amino acid sequence (Ile47 to Phe61) shared amongst all nephrozoan UBA2 orthologues (Fig. 3d). Olsen et al. showed that variants of residues (Asn56Ala, Leu57Ala, Arg59Ala) of this element result in loss of UBA2 function, and found that the very residue mutated in individual I14, Asp50, forms hydrogen bonds with Asn177 and Thr178 essential for proper UBA2 folding and thus its function (PS3) (Olsen et al. 2010). The variant was also absent in the databases (PM2) and predicted to be pathogenic by MutationTaster (PP3). Hence, we classified these variants as likely pathogenic according to the ACMG’s guidelines (1PS + 1PM + 2PP).
Novel candidate genes
Our analysis also revealed several novel, high-confidence candidate genes associated with limb defects. In individual I15 featuring severe mirror image foot polydactyly, we found a de novo frameshift variant in the gene encoding the high mobility group box 1 protein (HMGB1) (Supplementary Fig. 8). NM_002128.7(HMGB1):c.551_554delAGAA;p.(Lys184Argfs*44) leads to the replacement of the protein’s entire C-terminal 30-residue acidic tail by 41 other unrelated residues. The tail is normally formed by an Asp/Glu-repeat element, which is highly conserved among HMGB1 orthologues. This repeat element stabilizes HMGB1’s secondary structure and is crucial for its DNA-bending capacity (Belgrano et al. 2013; Anggayasti et al. 2020). The variant is not only absent from the databases but also no variant listed in gnomAD contains an amino acid residue except Glu or Asp in the acidic tail domain. HMGB1 has a pLI score of 1. In mouse and zebrafish studies, HMGB1 has been shown to regulate digit number during embryonic limb development by interacting with WNT, BMP, and SHH (Itou et al. 2011). We, therefore, consider HMGB1 to be a novel candidate gene for mirror image foot polydactyly.
Individual I16, who featured short stature, absent distal phalanges of the 5th fingers and toes, and dysplastic middle phalanges of the toes carried a de novo missense variant in the gene encoding semaphorin 3D (SEMA3D) (Supplementary Fig. 9). NM_152754.3(SEMA3D):c.1918G > A;p.(Asp640Asn) is absent from the 1000 Genomes Project database and is listed only 4 times in the Genome Aggregation Database (gnomAD). The Asp640 residue in the immunoglobulin-like domain of SEMA3D is highly conserved amongst vertebrates. The variant is predicted to be pathogenic by MutationTaster. SEMA3D regulates neural crest cell differentiation and is involved in the organogenesis of the heart (Sanchez-Castro et al. 2015), parathyroid gland (Singh et al. 2019), and, notably, limbs (Govindan et al. 2016). We, therefore, consider it a candidate gene for short stature with limb abnormalities.
In individual I17 we identified a paternally inherited frameshift variant in the aldehyde dehydrogenase 1 family member A2 gene (ALDH1A2) encoding retinaldehyde dehydrogenase 2 (Supplementary Fig. 10). Both, the patient and her father feature isolated cutaneous syndactyly of the fingers III–IV and the toes II–III. The variant NM_003888.4(ALDH1A2):c.35delT;p.(Val12Glyfs*31) is absent from the databases and is predicted to be disease-causing by MutationTaster. ALDH1A2 is a direct target of HOXA13 and plays a key role in vertebrate digit development by regulating, in particular, interdigital programmed cell death (Shou et al. 2013). Rescued ALDH1A2 knockout mice show reduced interdigital cell death and thus impaired digit separation during limb development resulting in syndactyly (Zhao et al. 2010). It is, therefore, likely that ALDH1A2:c.35delT caused the phenotype of syndactyly in individual I17 and her father.
Identification of noncoding variants
So far, the interpretation of disease-related variation has been focused on protein-coding DNA and the identification of variants that directly result in the disruption of specific gene functions.
Here, we aimed to develop a framework to prioritize a large number of noncoding variants from GS studies, by combining mouse genetic and human functional epigenetic data with in vivo-validated enhancer sequences. We then defined a limb-specific regulome that we used to filter all noncoding variants (Materials and Methods). Our potentially disease-relevant limb-specific regulome consists of 5,591,007 sites covering in total 7,294,220 bp, i.e. 0.24% of the human genome (hg19).
Overall, we identified 19,362 rare noncoding SNVs in the 69 index patients, of which 143 were located within the limb regulome (Fig. 4). First, we focused on the de novo variants and identified 6 calls located in potential regulatory elements. Two variants were excluded because they were called in cases with (likely) pathogenic coding or structural variants.

Pipeline of noncoding data analysis
Individual I18 presenting with bilateral syndactyly of fingers II-V featured the de novo call chr1:41948304AAG > A in intron 2 of EDN2. The position shows increased acetylation of H3K27 in human limb buds. The 2 bp deletion also removes one element of a 6-AG-repeat whose length is not conserved in vertebrates. Furthermore, EDN2 encodes endothelin 2, a potent vasoconstrictor with no evident link to limb development.
Individual I19 showing upper limb amelia featured three calls (chr5:157285900CACGTGGG > C, chr5:157285909CTCGG > C, chr5:157285915CACAACTG > C) referring to the same indel in intron 1 (15 bp downstream of the first exon–intron boundary) of CLINT1. However, CLINT1 shows only a moderate pLI score (0.54) and there is no evidence other than increased H3K27ac marks of its promoter region in human limb buds linking it to limb development.
We were not able to identify any rare variants in validated VISTA enhancers that showed enhancer activity in the limb bud.
Next, we focused on noncoding variants that were located close to one another in more than one case. In total, 3425 rare noncoding variants in the unsolved cases were positioned 300 bp or less apart from a variant in another unsolved case. 16 of these calls were located within the limb regulome, but in five of these variants, the other variant was positioned outside of the limb regulome.
In two cases both variants were positioned within the limb regulome and within 300 bp: individual I21 and individual I22, both showing finger syndactyly, harbored the overlap** deletions chr22:24552064GGGGGCCGGGACTGGGGCCGGGACT > G and chr22:24552086ACTGGGGCCGGGG > A, respectively. The deletions are positioned in intron 29 of CABIN1, in an evolutionarily partially conserved element, that shows increased H3K27ac marks in human embryonic limb buds. However, both deletions were inherited from unaffected parents.
Eight of the close variants were double hits (i.e. we detected rare calls not in just one but two index patients at four positions of the potential limb regulome). However, none of these four pairs of index patients showed overlap** phenotypes.
We identified no coding variant of a known limb disease gene in trans with a conserved, rare noncoding variant of the same TAD.
In summary, despite extensive efforts, we were not able to identify any noncoding SNVs that showed convincing evidence to be causal in congenital limb malformations.
Discussion
In this study, we set out to determine the potential of GS as a comprehensive diagnostic tool to determine all kinds of genetic variants associated with congenital limb malformation.
In our cohort of patients with congenital limb malformations, GS was able to detect both previously described and novel causative genetic variants in already established limb malformation associated genes. In addition, it enabled the identification of three candidate genes and the independent verification of the novel disease gene UBA2 for causing ectrodactyly (Yamoto et al. 2019). Our approach was able to detect SNVs and structural variants. Finally, GS proved to be a powerful strategy to identify genomic variants previously missed by most other approaches, including repeat expansions and complex structural variants. In total, we identified variants that we consider to be likely pathogenic/disease-associated in 12 of 69 cases (17.4%). This diagnostic yield is comparable to the recent landmark study conducted by the British National Health Service that used GS in cohorts with other congenital disease entities (Turro et al. 2020). A clear advantage of GS compared to most other technologies is the ability to detect copy number neutral variants and to gain position information on CNVs. In our cohort of only 69, we were able to detect two complex variants, an inversion at the FGF8 locus and a translocated triplication including the SHH gene. Both were missed by standard technologies. Further research is necessary to clarify their exact pathomechanisms. The variants identified in the genes HMGB1, SEMA3D and ALDH1A2 are all likely to cause loss of function. The genes were previously associated with vertebrate limb development in animal studies and the variants either arose de novo or segregate with the respective phenotype. However, we could not identify unrelated individuals featuring comparable variants in these candidate genes and similar phenotypes. Future research is necessary to identify such to establish the described candidates as disease genes. Our findings once again highlight the role of GS as an attractive one-test-for-all strategy for clinically very heterogeneous cohorts such as congenital malformation syndromes or intellectual disability (Gilissen et al. 2014; Turro et al. 2020). The total cost of the various conventional tests currently used in clinical routine far exceeds that of trio GS.
One of the main challenges of GS data is the medical interpretation of changes in the noncoding DNA. While most clinical GS studies tend to ignore noncoding SNVs (Gilissen et al. 2014) there are recent anecdotal reports of noncoding variants as the cause of Mendelian disorders (Lettice et al. 2003; Jeong et al. 2008; Albers et al. 2012; Bhatia et al. 2013; Weedon et al. 2014; Bae et al. 2014), although there is no established systematic approach, yet. Therefore, we set out to develop a framework to prioritize such noncoding variants associated with congenital limb malformation. We used a combinatorial approach of mouse and human epigenetic data, in vivo validated enhancer sequences, knock-out mice, and the recent knowledge about 3D genome folding, and the cis-regulatory architecture of the genome to define a limb regulome. This limb regulome consists of 0.24% of the genome and includes all known in vivo-validated limb enhancer elements. Contrary to our expectation, we could only identify candidate loci, but no definitely pathogenic noncoding variants. These findings are in stark contrast to our recent study where we demonstrated that CNVs affecting noncoding regulatory elements are a major cause of congenital limb malformations (Flöttmann et al. 2018).
While our results suggest that GS is sensitive to classical sequence variants, it is noteworthy that the method cannot detect epigenetic variants. Epimutations (e.g. imprinting defects) are known to cause inheritable human disease. However, to our knowledge, no epimutation has been linked to congenital limb malformation yet.
Tools for the analysis of GS data are continuously being developed further and the precision of algorithms to call structural variants can certainly be improved. We expect the diagnostic rate to increase steadily with the accuracy of the instruments invoked to analyze GS data.
Code and data availability
Code and data (pseudonymized, grouped where possible, and minimized) are available upon reasonable request.
References
Abe KT, Rizzo IMPO, Coelho ALV et al (2018) 19q13.11 microdeletion: clinical features overlap** ectrodactyly ectodermal dysplasia-clefting syndrome phenotype. Clin Case Rep 6:1300–1307. https://doi.org/10.1002/ccr3.1600
Aerden M, Bauters M, Van Den Bogaert K et al (2020) Genotype-phenotype correlations of UBA2 mutations in patients with ectrodactyly. Eur J Med Genet 63:104009. https://doi.org/10.1016/j.ejmg.2020.104009
Albers CA, Paul DS, Schulze H et al (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44(435–9):S1-2. https://doi.org/10.1038/ng.1083
Anggayasti WL, Ogino K, Yamamoto E et al (2020) The acidic tail of HMGB1 regulates its secondary structure and conformational flexibility: a circular dichroism and molecular dynamics simulation study. Comput Struct Biotechnol J 18:1160–1172
Bae B-I, Tietjen I, Atabay KD et al (2014) Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 343:764–768. https://doi.org/10.1126/science.1244392
Belgrano FS, de Abreu da Silva IC, Bastos de Oliveira FM et al (2013) Role of the acidic tail of high mobility group protein B1 (HMGB1) in protein stability and DNA bending. PLoS ONE 8:e79572. https://doi.org/10.1371/journal.pone.0079572
Berisha SZ, Shetty S, Prior TW, Mitchell AL (2020) Cytogenetic and molecular diagnostic testing associated with prenatal and postnatal birth defects. Birth Defects Res 112:293–306. https://doi.org/10.1002/bdr2.1648
Bhatia S, Bengani H, Fish M et al (2013) Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet 93:1126–1134. https://doi.org/10.1016/j.ajhg.2013.10.028
Brison N, Debeer P, Tylzanowski P (2014) Joining the fingers: a HOXD13 story. Dev Dyn 243:37–48. https://doi.org/10.1002/dvdy.24037
Cao J, Spielmann M, Qiu X et al (2019) The single-cell transcriptional landscape of mammalian organogenesis. Nature 566:496–502. https://doi.org/10.1038/s41586-019-0969-x
Chowdhury S, Bandholz AM, Parkash S et al (2014) Phenotypic and molecular characterization of 19q12q13.1 deletions: a report of five patients. Am J Med Genet A 164A:62–69. https://doi.org/10.1002/ajmg.a.36201
Cotney J, Leng J, Yin J et al (2013) The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154:185–196. https://doi.org/10.1016/j.cell.2013.05.056
de Mollerat XJ, Gurrieri F, Morgan CT et al (2003) A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 12:1959–1971
Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542:433–438. https://doi.org/10.1038/nature21062
Dixon JR, Selvaraj S, Yue F et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380. https://doi.org/10.1038/nature11082
Flöttmann R, Knaus A, Zemojtel T et al (2015) FGFR2 mutation in a patient without typical features of Pfeiffer syndrome—the emerging role of combined NGS and phenotype based strategies. Eur J Hum Genet 58:376–380. https://doi.org/10.1016/j.ejmg.2015.05.007
Flöttmann R, Kragesteen BK, Geuer S et al (2018) Noncoding copy-number variations are associated with congenital limb malformation. Genet Med 20:599–607. https://doi.org/10.1038/gim.2017.154
Gilissen C, Hehir-Kwa JY, Thung DT et al (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature 511:344–347. https://doi.org/10.1038/nature13394
Govindan J, Tun KM, Iovine MK (2016) Cx43-dependent skeletal phenotypes are mediated by interactions between the Hapln1a-ECM and Sema3d during Fin regeneration. PLoS ONE 11:e0148202. https://doi.org/10.1371/journal.pone.0148202
Holder-Espinasse M, Jamsheer A, Escande F et al (2019) Duplication of 10q24 locus: broadening the clinical and radiological spectrum. Eur J Hum Genet 27:525–534. https://doi.org/10.1038/s41431-018-0326-9
Holtgrewe M, Stolpe O, Nieminen M et al (2020) VarFish: comprehensive DNA variant analysis for diagnostics and research. Nucleic Acids Res 48:W162–W169. https://doi.org/10.1093/nar/gkaa241
Itou J, Taniguchi N, Oishi I et al (2011) HMGB factors are required for posterior digit development through integrating signaling pathway activities. Dev Dyn 240:1151–1162
Jeong Y, Leskow FC, El-Jaick K et al (2008) Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat Genet 40:1348–1353. https://doi.org/10.1038/ng.230
Klopocki E, Lohan S, Doelken SC et al (2012) Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet 49:119–125. https://doi.org/10.1136/jmedgenet-2011-100409
Kroeldrup L, Kjaergaard S, Kirchhoff M et al (2012) Duplication of 7q36.3 encompassing the Sonic Hedgehog (SHH) gene is associated with congenital muscular hypertrophy. Eur J Med Genet 55:557–560. https://doi.org/10.1016/j.ejmg.2012.04.009
Lettice LA, Heaney SJH, Purdie LA et al (2003) A long-range Shh enhancer regulates expression in the develo** limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12:1725–1735. https://doi.org/10.1093/hmg/ddg180
Levy SE, Myers RM (2016) Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet 17:95–115. https://doi.org/10.1146/annurev-genom-083115-022413
Lindstrand A, Eisfeldt J, Pettersson M et al (2019) From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med 11:68. https://doi.org/10.1186/s13073-019-0675-1
Lohan S, Spielmann M, Doelken SC et al (2014) Microduplications encompassing the Sonic hedgehog limb enhancer ZRS are associated with Haas-type polysyndactyly and Laurin-Sandrow syndrome. Clin Genet 86:318–325. https://doi.org/10.1111/cge.12352
Muenke M, Schell U, Hehr A et al (2014) A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet 8:269–274. https://doi.org/10.1038/ng1194-269
Nieminen M, Stolpe O, Schumann F et al (2020) SODAR core: a Django-based framework for scientific data management and analysis web apps. J Open Source Softw JOSS 55:1584. https://doi.org/10.21105/joss.01584
Olsen SK, Capili AD, Lu X et al (2010) Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463:906–912. https://doi.org/10.1038/nature08765
Sanchez-Castro M, Pichon O, Briand A et al (2015) Disruption of the SEMA3D gene in a patient with congenital heart defects. Hum Mutat 36:30–33. https://doi.org/10.1002/humu.22702
Shou S, Carlson HL, Perez WD, Stadler HS (2013) HOXA13 regulates Aldh1a2 expression in the autopod to facilitate interdigital programmed cell death. Dev Dyn 242:687–698. https://doi.org/10.1002/dvdy.23966
Singh A, Mia MM, Cibi DM et al (2019) Deficiency in the secreted protein Semaphorin3d causes abnormal parathyroid development in mice. J Biol Chem 294:8336–8347. https://doi.org/10.1074/jbc.RA118.007063
Turro E, Astle WJ, Megy K et al (2020) Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583:96–102. https://doi.org/10.1038/s41586-020-2434-2
Visel A, Blow MJ, Li Z et al (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457:854–858. https://doi.org/10.1038/nature07730
Weedon MN, Cebola I, Patch A-M et al (2014) Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 46:61–64. https://doi.org/10.1038/ng.2826
Xue Y, Ankala A, Wilcox WR, Hegde MR (2015) Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 17:444–451. https://doi.org/10.1038/gim.2014.122
Yamoto K, Saitsu H, Nishimura G et al (2019) Comprehensive clinical and molecular studies in split-hand/foot malformation: identification of two plausible candidate genes (LRP6 and UBA2). Eur J Hum Genet 27:1845–1857. https://doi.org/10.1038/s41431-019-0473-7
Zhao X, Brade T, Cunningham TJ, Duester G (2010) Retinoic acid controls expression of tissue remodeling genes Hmgn1 and Fgf18 at the digit-interdigit junction. Dev Dyn 239:665–671
Acknowledgements
We would like to thank all families for their collaboration and contribution to this project. J.E. is supported by a scholarship of the Studienstiftung des deutschen Volkes e.V. M.A.M. is a participant in the BIH Charité Digital Clinician Scientist Program founded by the late Prof. Duska Dragun and funded by the Charité-Universitätsmedizin Berlin and the Berlin Institute of Health.
Funding
Open Access funding enabled and organized by Projekt DEAL. A.J. was supported by the grant from the Polish National Science Centre UMO-2016/22/E/NZ5/00270. M.S. is supported by grants from the Deutsche Forschungsgemeinschaft (DFG) (SP1532/3-1, SP1532/4-1 and SP1532/5-1), the Max Planck Foundation and the Deutsches Zentrum für Luft- und Raumfahrt (DLR 01GM1925).
Author information
Authors and Affiliations
Contributions
Jonas Elsner, Martin A. Mensah, Stefan Mundlos and Malte Spielmann designed the study. Jonas Elsner, Martin A. Mensah, Manuel Holtgrewe, Jakob Hertzberg and Marie Coutelier performed the variant filtration and processed data. Jonas Elsner, Martin A. Mensah and Malte Spielmann investigated and together with Stefan Mundlos and Denise Horn critically evaluated variants. Martin A. Mensah, Stefania Bigoni, Andreas Busche, Deepthi C. de Silva, Nursel Elçioglu, Isabel Filges, Erica Gerkes, Katta Girisha, Luitgard Graul-Neumann, Jane Hurst, Aleksander Jamsheer, Peter Krawitz, Ingo Kurth, Susanne Markus, Andre Megarbane, André Reis, Miriam Reuter, Daniel Svoboda, Christopher Teller, Beyhan Tuysuz, Seval Türkmen, Meredith Wilson, Rixa Woitschach, Inga Vater, Almuth Caliebe, Wiebke Hülsemann, Denise Horn, Stefan Mundlos and Malte Spielmann examined the patients and provided samples. Jonas Elsner and Martin A. Mensah visualised the workflow and results. Jonas Elsner, Martin A. Mensah, Stefan Mundlos and Malte Spielmann wrote and reviewed the manuscript with the approval of all authors. Stefan Mundlos and Malte Spielmann supervised the project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
This study was approved by the ethics committees of the Charité-Universitätsmedizin Berlin (EA2/087/15) and the Medical Chamber (Ärztekammer) Hamburg and conducted in accordance with the declaration of Helsinki. The Charité’s ethic commission served as the central IRB.
Consent to participate
Participants or their legal guardians provided written informed consent for participation in this study. Consents of all participants were archived. All published data has been pseudonymized, grouped where possible, and minimized.
Consent for publication
Participants or their legal guardians provided written informed consent for pseudonymized publication of their data. Consents of all participants were archived. All published data have been pseudonymized, grouped where possible, and minimized.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.



Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Elsner, J., Mensah, M.A., Holtgrewe, M. et al. Genome sequencing in families with congenital limb malformations. Hum Genet 140, 1229–1239 (2021). https://doi.org/10.1007/s00439-021-02295-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02295-y