
Abstract
During their intrathymic development, nascent T cells are empowered to protect against pathogens and to be operative for a life-long acceptance of self. While autoreactive effector T (Teff) cell progenitors are eliminated by clonal deletion, the intrathymic mechanisms by which thymic regulatory T cell (tTreg) progenitors maintain specificity for self-antigens but escape deletion to exert their regulatory functions are less well understood. Both tTreg and Teff development and selection result from finely coordinated interactions between their clonotypic T cell receptors (TCR) and peptide/MHC complexes expressed by antigen-presenting cells, such as thymic epithelial cells and thymic dendritic cells. tTreg function is dependent on expression of the FOXP3 transcription factor, and induction of FOXP3 gene expression by tTreg occurs during their thymic development, particularly within the thymic medulla. While initial expression of FOXP3 is downstream of TCR activation, constitutive expression is fixed by interactions with various transcription factors that are regulated by other extracellular signals like TCR and cytokines, leading to epigenetic modification of the FOXP3 gene. Most of the understanding of the molecular events underlying tTreg generation is based on studies of murine models, whereas gaining similar insight in the human system has been very challenging. In this review, we will elucidate how inborn errors of immunity illuminate the critical non-redundant roles of certain molecules during tTreg development, shedding light on how their abnormal development and function cause well-defined diseases that manifest with autoimmunity alone or are associated with states of immune deficiency and autoinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Regulatory T cells (Treg) represent a specific T cell subset exercising a primary role in multiple essential immune functions. They regulate immune responses to self and foreign antigens, and control inflammation and immune surveillance. Regulatory T cells suppress the activation, expansion, and effector functions of the T cells that mediate immune responses following pathogen or alloantigen recognition. There are several types of regulatory T cells but only the naturally occurring CD4+ CD25hi FOXP3+ thymic Treg (tTreg) differentiate in the thymus as part of T cell ontogeny [1,2,3,4]. Induced Treg (iTreg) can be generated from CD4+CD25− cells in vitro in both mouse and human in the presence of TGFβ; however, human iTreg are not suppressive, suggesting that mouse and human Treg have different differentiation requirements [5]. FOXP3 is a master transcriptional regulator that controls the tTreg differentiation program, is essential for tTreg function, and its constitutive expression distinguishes tTreg from other cells, e.g., activated Teff, that transiently express FOXP3. A subset of Treg with a comparable phenotype (CD4+ CD25hi FOXP3+) has also been demonstrated in the mouse to differentiate in peripheral (i.e., extrathymic) tissues and is hence referred to as peripheral Treg (pTreg). Whether pTreg are also present in humans is uncertain since their identification is difficult because definitive markers that distinguish between the two subsets are presently missing [6]. Type 1 regulatory T cells (Tr1) are another peripherally induced CD4+ Treg population, which, in notable contrast to tTreg, develops and exerts its suppressive functions independent of FOXP3. Tr1 cells are also involved in maintaining immune tolerance and the balance between effector and suppressive activities, as recently reviewed elsewhere [7]. The occurrence of multi-organ autoimmunity, early in life, in patients with FOXP3 mutations demonstrate that tTreg are non-redundant, and that no other regulatory population, e.g., Tr1 or pTreg, can complement loss of FOXP3+ tTreg. This review will focus on tTreg and their early development, and will thus describe those intrathymic events that forge the cells’ unique identity under both physiological and prototypical disease conditions.
Since first described in the late 1990s in mice, a consistent body of knowledge has not only characterized the identity of tTreg but also described many, if not all, of the mechanisms by which these cells suppress Teff functions [8]. The precise molecular mechanisms that dictate tTreg differentiation, control the correct spatio-temporal manifestations of this complex developmental process, and thus enable a divergence from Teff differentiation remain, however, still largely undefined. Gaining a detailed understanding of tTreg development has been made possible by studying murine models, whereas comparable and complementary investigations in humans have been significantly more difficult to attain [9]. Therefore, most of our understanding of human thymopoiesis and specifically the role of the thymic microenvironment in this process has been extrapolated from mice with either spontaneously occurring or engineered loss and gain of gene functions. These studies take advantage of the ability to study embryonic thymic development or to trace cell lineages, which are not easily possible in human studies. However, postnatal thymopoiesis and tTreg progenitors can be studied in thymic tissue obtained from infants, children, and adults who undergo cardiac surgery, either for the correction of congenital heart disease (CHD) or for a heart transplant where the surgical procedures dictate the removal of thymic lobes. Consistent with primary immune deficiencies being informative “experiments of nature” to gain further insight into the immune development and function, the study of thymic tissue from genetic disorders affecting tTreg development, e.g., the chromosome 22q11.2 deletion syndrome (22q11.2DS) (discussed below and elsewhere in this issue), can be used as disease models analogous to murine models with defined genetic abnormalities [10]. However, the ability to study fetal development in human model diseases is limited and genetic heterogeneity can create additional confounders that are more easily controlled in murine models. Another approach to studying human thymopoiesis is chimeric mouse models transplanted with human hematopoietic stem and progenitor cells (HSPC). These models rely on interactions of the human HSPC with either the native murine microenvironments, co-transplanted human microenvironmental cells (e.g., human marrow and/or thymus), or “humanized” murine microenvironments that have been genetically altered to express human genes relevant to study hematopoiesis and immune functions. To date, the robustness of these chimeric mouse models is still limited by species-specific differences in cytokine ligand-receptor pairs and incomplete humanization of the murine MHC so that T lymphocyte differentiation, including that of Tregs, does not fully mirror human thymopoiesis [11]. A third approach to probe human T cell development uses thymus organoids, which have been developed to study in vitro early events in human thymopoiesis. To date, these platforms have not been used to examine intrathymic tTreg differentiation, selection, and maturation [12, 13]. While strategies to either expand tTreg or convert Teff into tTreg-like cells are the focus of intense research on the use of Treg as a cellular therapy, these in vitro or in vivo protocols may not be related to the normal development of tTreg [14, 15].
In this review, we will summarize the current knowledge of human tTreg identity and function, the role of thymic mechanisms that determine tTreg output, and the molecular and cellular basis for genetic diseases of tTreg function that lead to early-onset autoimmunity. Insights gained from these rare genetic diseases can be extended to improve the understanding of more common autoimmune diseases.
tTreg identity and mechanisms of function
The phenotypic discovery and functional characterization of murine CD4+ CD25hi tTreg by Sakaguchi et al. in 1995 was subsequently extended to human peripheral blood where a corresponding phenotype was found [16,17,18,19]. Human CD4+ CD25hi tTreg described originally were themselves anergic, yet able to suppress the proliferation of naïve and memory effector T cells. They could also be stimulated to proliferate in vitro under specific short-term culture conditions [17,18,19]. Human tTreg represent a small (3–8%) subpopulation among peripheral CD4+ T cells. The number of tTreg is increased early in life in parallel with the higher lymphocyte count observed in infants, although the proportion of tTreg within the lymphocyte compartment remains relatively constant into adulthood [20]. Analyses of human T cell differentiation from fetal vs adult HSPC in chimeric mice indicate an inherent bias of fetal progenitors towards a Treg fate [21]. As in the mouse, human tTreg can be identified immunophenotypically by a constitutive high-level expression of the α chain of the IL-2 receptor (IL2R, CD25), and the lack of alpha chain of the IL-7 receptor (CD127) expression [3]. CD25 expression alone is an unreliable marker of tTreg because it is also expressed by activated Teff cells, albeit at lower levels. Because most Teff also express CD127, the combination of high CD25 and absent CD127 expression can distinguish tTreg from activated CD4 T cells [22]. Other functionally important surface markers of human tTreg include the cytotoxic T lymphocyte antigen 4 (CTLA4) and the Lymphocyte Activation Gene-3 (LAG-3). Importantly, the vast majority of CD4+ CD25hi CD127− cells (> 80%) express the transcription factor FOXP3 [23].
The constitutively high expression of nuclear FOXP3 is not only a phenotypic marker of tTreg, but is also necessary to establish the cells’ regulatory activity [23]. FOXP3 is a member of the forkhead (Fkh)-winged helix family of transcription factors, encoding a protein with a nuclear localization signal, a zinc finger, and leucine zipper–containing region, and an Fkh domain, all of which are necessary for function [24, 25]. Alternative splicing of human FOXP3 can generate 4 different isoforms (full length, Δ2, Δ7, and Δ2 Δ7, lacking exon 2, exon 7, or both exons 2 and 7) [26,27,28,29]. The most abundantly expressed human FOXP3 isoforms are the full length and the Δ2. The exon 2 encodes for the proline-rich domain and exon 7 contains the leucine zipper domain of FOXP3. The splice variants lacking exon 2 appear to be at least partially functional and to protect from severe IPEX syndrome [26,27,28,29,30], whereas the isoform lacking exons 2 and 7 is unable to confer suppressive function [30, 31].
One mechanism by which FOXP3 operates as a master regulator of T cell function is through direct interactions with genes containing the Fkh consensus binding sequence, GTAAACA. This recognition requires both the zinc finger/leucine zipper region and Fkh protein domains, as mutations in either abrogate FOXP3 function and result in autoimmunity [25]. At least 700–1400 genes are directly bound by the FOXP3 protein. However, the number of genes directly recognized by FOXP3 is estimated to be less than 10% that of the number of genes whose expression is regulated by FOXP3 [32]. This difference is explained by either activation of secondarily regulated genes by FOXP3-bound transcription factor genes or interactions of FOXP3 with other proteins, e.g., epigenetic modifiers that alter gene expression [25, 32]. Interacting transcription factor genes include those encoding NFAT, RUNX1/AML1, IRF4, GATA3, REL, and RORγT [25]. A recent study has demonstrated that many murine tTreg-associated genes are controlled by non-tTreg-specific transcription factors like TCF1, expression of which is modulated by FOXP3 [33]. The intermediary proteins have intrinsic capacity to bind to FOXP3-regulated genes, but the formation of macromolecular complexes with FOXP3 either potentiates or possibly stabilizes the effects of these other transcription factors. Examples of FOXP3 protein–protein interactions are the recruitment of DNA- or histone-modifying enzymes that alter the epigenome of FOXP3 controlled sequences by changing DNA methylation or post-transcriptionally modifying histone methylation or acetylation [25]. For example, FOXP3-dependent expression of the Ikaros family member Eos leads to the epigenetic silencing of the IL-2 gene in tTreg [24, 34]. The central observation, for which our mechanistic understanding remains incomplete, is that the combined effects of the direct and indirect gene activation and repression confer a stable regulatory phenotype on FOXP3-expressing tTreg.
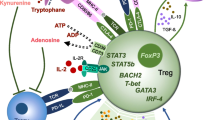
Mechanisms of tTreg-mediated immune regulation
Immune regulation by tTreg depends on their ability to suppress the activity of Teff [35]. Much of the suppressive activity depends on cell–cell contact, indicating the important role of tTreg surface proteins. The LAG-3 expressed by tTreg binds to HLA-DR, thereby inhibiting the T cell receptor (TCR)-mediated activation of CD4+ Teff [36]. The binding of CTLA4 to CD80 and CD86 on APC blocks their interactions with CD28 expressed by Teff and thereby suppresses co-stimulation [37]. CTLA4 also upregulates indoleamine 2,3-dioxygenase (IDO) expression by dendritic cells (DC), which starves the T cells by depletion of tryptophan [38]. Perforin-mediated cytolysis of target cells by tTreg has been described as another mechanism of suppression [39]. Resting tTreg cells are typically hypoproliferative but, like activated Teff, are able to undergo IL-2-driven expansion in vivo; this property has been used in vitro to generate cell products that have been used for therapeutic purposes [14, 19]. Although tTreg do not produce IL-2 themselves, their expression of the CD25 subunit of the IL2R confers high-affinity binding of IL-2. Consequently, tTreg have a proliferative advantage in IL-2 responsiveness over other T cell populations [40,41,42]. Besides selectively promoting the growth of tTreg in response to IL-2, the high-affinity IL-2 receptor formed on tTreg by their high-level CD25 expression also scavenges IL-2 from their local milieu, thereby depriving Teff of this major T cell growth factor. Reflecting their indispensable role in Treg biology, the experimental loss of function of prototypical tTreg markers like CTLA4 or CD25 causes loss of tTreg function in mice, resulting in lymphoproliferative diseases accompanied by various degrees of autoimmunity [9]. In addition to IL-2 scavenging, there are other suppressive functions imposed by tTreg that are not cell contact dependent. For example, tTregs express the ectoenzymes CD39 and CD73, which generate immunosuppressive purine nucleosides [43]. The tTregs produce limited amount of cytokines, at least in vitro, and those include immunoregulatory cytokines such as IL-10, TGFβ, and IL-35, which can suppress some Teff activities [6].
The relative balance of suppressive mechanisms used by individual tTreg may depend on target cells and tissue milieu. Mature tTreg can undergo further differentiation in the periphery or may modulate their gene expression profile in response to activation state, distinctive milieus, or interactions with different types of Teff [44]. Like Teff, tTreg can also be subdivided by activation state. Resting and activated tTreg can be distinguished by expression of the CD45RA isoform in resting naïve tTreg and lack of CD45RA on activated tTreg [45]. Markers such as LAG3, TIGIT, PD-1, GARP, and TNFRSF18 (GITR) are expressed by or upregulated by tTreg undergoing differentiation to an activated state where they exert maximal suppressive function. These proteins are also found on peripherally induced pTreg [46]. Peripheral trafficking of tTreg into different tissues and continued encounter with multiple environmental factors further increase the cells’ heterogeneity [46]. Interestingly, different types of tTreg have been described in different tissues and in response to Th1, Th2, and Th17 subsets of Teff, suggesting that tTreg either adapt to their local milieu, or that specific tTreg subsets selectively traffic to or are selected by individual peripheral microenvironments [6, 47]. The ability to fully characterize human tTreg subsets and draw their differentiation trajectory remains limited in humans, although single-cell analyses are likely to at least partly redress these problems [48].
Regulation of FOXP3 expression
Given the central role of FOXP3 in tTreg differentiation and function, understanding the mechanisms that regulate its expression has been critical for understanding tTreg biology. The FOXP3 gene promoter is relatively weak, but is activated by binding to NFAT, AP-1, and the cyclic AMP-response element-binding protein (CREB)–activating protein 1 (ATF1), which are all downstream of TCR signaling, as well as two different forkhead family proteins, FOXO1 and FOXO3, which are downstream of CD28 co-stimulation and other pathways [25, 49, 50]. tTreg have a metabolic profile distinct from that of Teff, in which they use fatty acid oxidation (FAO) and pyruvate-dependent oxidative phosphorylation (OXPHOS) as their energy source [51]. By-products of these pathways, such as phosphoenol pyruvate (PEP) and mitochondrial reactive oxygen species (ROS), are predicted to increase NFAT activation after TCR signaling, which could increase FOXP3 expression [51].
Because of the weak promoter, high-level FOXP3 expression depends on three conserved non-coding sequences (CNS) that function as intronically located enhancer elements. CNS1 and CNS2 are in the first intron between non-coding exons -2b and -1, and CNS3 is in the second intron between coding exons 1 and 2 of the murine and human FOXP3 genes [24, 25]. The distal CNS3 enhancer contains response elements for the NF-κB REL protein, which binds after TCR-mediated NF-κB activation. Together, the activation of the FOXP3 promoter and CNS3 by TCR signaling to initiate FOXP3 expression explain the expression of FOXP3 by activated Teff. Although TCR signaling alone is sufficient to induce FOXP3 expression in mature human Teff, this is transient, as constitutive expression requires the tTreg-specific or pTreg-specific activities of CNS2 and CNS1, respectively. The transient expression of FOXP3 by activated Teff, along with their activation induced CD25 expression, can lead to immunophenotypic confusion with tTreg, which constitutively express both FOXP3 and CD25. It is not known whether transient expression of FOXP3 by Teff is functional, e.g., putting a brake on Teff expansion. Phenotypically, tTreg can still be distinguished from activated Teff by their higher levels of CD25 expression and absence of CD127, as detailed above. CNS1, the most promoter proximal enhancer element, is important in the development of extrathymic pTreg. In these cells, FOXP3 expression is induced via transcriptional activation resulting from binding of the TGFβ-related signaling protein SMAD3, NFAT, and retinoic acid receptor-retinoid X receptor (RAR-RXR) [25].
Stable FOXP3 gene expression in tTreg is enabled epigenetically by the demethylation of a specific CpG island in CNS2 [52, 53]. Also known as Treg-specific demethylated region (TSDR), its epigenetic modification occurs exclusively, at least in humans, during tTreg differentiation, stabilizes the tTreg’s functional identity, and is maintained throughout the cell’s life, thus serving as a suitable molecular landmark. The demethylation of CNS2 allows binding by a number of transcription factors, including the aforementioned REL and CREB-ATF1, as well as RUNX1-core-binding factor subunit-β (CBFβ), ETS1, STAT5, and FOXP3 itself [25]. Since STAT5 is activated by IL2R signaling, the high-level expression of the CD25 subunit, along with the positive transcriptional effects of FOXP3 itself, generate feed-forward mechanisms for continued FOXP3 gene expression. Further stabilization of FOXP3 expression is provided by the recruitment of TET family demethylases by STAT5, thereby ensuring continuation of the demethylated state and accessibility of CNS2 to the transcription factors [54,55,56]. The protein–protein interactions, the necessity vs redundancy, and the temporal sequence of binding of the transcription factors that control FOXP3 expression in human tTreg are not well characterized, but the overall effect is to stabilize FOXP3 expression regardless of the cellular environment or activation state. The tTreg-specific demethylation of CNS2 is a unique epigenetic mark that allows for precise and unbiased quantification of tTreg among peripheral blood lymphocytes [57].
Several critical immunoregulatory pathways contribute to the maintenance of the tTreg phenotype by promoting FOXP3 expression. Besides the growth advantage conferred by the IL-2R, its downstream activation of STAT5 results in recruitment of TET-demethylating enzymes to the TSDR, thereby creating a positive feedback loop that can reinforce FOXP3 expression [25, 54,55,56]. In addition to IL-2, IL15 has also been described as important for STAT5-mediated tTreg survival and proliferation in the thymus [58]. While FOXP3 expression by tTreg is generally stable, the pro-inflammatory cytokine IL-6 induces recruitment of DNMT1 methyltransferase to both the FOXP3 promoter and the TSDR, which can cause remethylation of the embedded CpG island and repression of FOXP3 expression [25, 54, 55]. This phenomenon is likely to contribute to the disruption of immune tolerance induced by IL-6.
Intrathymic tTreg development: a pathway divergent from that of Teff cells
Given the importance of TCR and cytokine (IL-2) signaling for tTreg function in prevention of autoimmunity and maintenance of immune homeostasis, understanding the intrathymic events underlying the formation of their TCR repertoire and acquisition of signaling competence has been a subject of great research interest. Experiments to elucidate this important process derive mainly from murine models, whereas the precise cellular conditions and molecular events required to assure normal human tTreg development remain still ill-defined [3, 9]. Besides expressing unique sets of genes critical to their suppressive functions, human tTreg have a defined TCR repertoire which, in healthy subjects, is distinct from that of the Teff cells, in line with what has been observed in mice [58]. However, in one study of human Tregs, the overlap between the two TCR repertoires is higher than that reported in the mouse, resulting in greater heterogeneity of the human tTreg repertoire than suppositionally predicted if they were only reactive to self-antigens [59].
Similar to the process for Teff development, the engagement of clonotypic TCR with the self-peptide/MHC complex, at the double positive (DP) and early CD4 single-positive (SP) stages, both drives further differentiation and expansion and acts as a selecting signal for intrathymic Treg development. Initial positive selection of DP thymocytes in the thymic cortex depends on expression of TCR with adequately high affinity for a peptide/MHC complex. Thymocytes with high-affinity TCR for self-ligands ectopically expressed in the thymic medulla are subsequently deleted there, those with low affinity become naïve Teff cells, and T cells that recognize self-antigens via an intermediate affinity TCR develop into tTreg cells [60]. In the mouse, clonal deletion also occurs in the thymic cortex, but this has not been confirmed in the human thymus [61]. In contrast to the mouse where intrathymic tTreg commitment is first observed in the single-positive population, human CD25+ cells with demethylation of CNS2 (see below) and a tTreg-like TCR repertoire have been noted among DP cortical thymocytes. This earlier tTreg commitment in human thymus could be explained if TCR activation, e.g., through the FOXP3 promoter and CNS3, is sufficient to initiate sustained FoxP3 expression [62].
Experiments in Nur77-GFP transgenic reporter mice have shown that Nur77-driven GFP expression is proportional to the TCR signaling strength. CD25+ tTreg progenitors had a significantly higher expression of Nur77-GFP than conventional CD4+ Foxp3− T cells, consistent with a higher degree of self-reactivity of the TCR expressed by tTreg cells. These findings confirm the importance of the TCR-peptide/MHC complex interactions in tTreg selection [63, 64]. Because the deletion of autoreactive Teff is leaky, some self-reactive T cells are present in the periphery [65, 66]. The experiments showing greater TCR signal strength in tTreg progenitors predicts that mature tTreg are ready to recognize self-antigens with a relatively higher affinity compared to Teff cells specific for the same self-antigens.
The signaling processes observed in mature tTreg after TCR engagement are thought to be similar to those induced in develo** thymocytes. Activation of NF-κB and resultant Rel binding to CNS3 and NFAT binding to the promoter of the FOXP3 gene are expected to lead to FOXP3 expression and subsequent induction of other FOXP3-dependent genes, e.g., CD25 and CTLA4 [25]. However, TCR signaling is also predicted to cause transient FOXP3 expression in non-tTreg. Hence, this signal does not explain the chromatin remodeling and epigenetic modifications, including demethylation, occurring at CNS2 (TSDR) to assure constitutive FOXP3 expression. Crucially, the identification of FOXP3+ thymocytes does not establish that all such cells are tTreg progenitors. The establishment of constitutive FOXP3 expression specifically in tTreg progenitors must depend on other processes besides TCR signaling. Interestingly, FOXP3 itself may alter TCR repertoire selection by tTreg progenitors. Using humanized mice transplanted with CD34+ hematopoietic stem cell progenitors in which FOXP3 expression was persistently downmodulated with an siRNA modification, we observed direct control of the strength of TCR signaling by FOXP3, which correlated with Nur77 expression [67]. This direct effect would further support the importance of persistent high expression of FOXP3 in tTreg development and selection of a self-reactive TCR repertoire. The altered signal strength could only alter selection of FOXP3+ tTreg progenitors in the thymic medulla, since positive selection of immature cortical thymocytes occurs prior to onset of FOXP3 expression. Consistent with this understanding is the observation that in Foxp3−/− mice, the TCR repertoire has a higher degree of overlap between tTreg and Teff, compared to that of wild-type tTreg and Teff [63].
Besides the importance of intrathymic TCR signaling in establishing the tTreg repertoire and initiating FOXP3 expression, cytokine signaling also plays a central role. IL-2 provides the survival and STAT5-mediated differentiation signals to CD25+ FOXP3− Treg progenitors, after which FOXP3 expression begins [68, 69]. IL-2-inducible phospho-STAT5 (pSTAT5) is also likely to be critical to FOXP3 transactivation by direct binding to CNS2, as well as by recruiting TET proteins to demethylate CNS2, thereby generating the TSDR essential for the establishment of constitutive FOXP3 expression [54,55,56]. Within the murine thymic medulla where both tTreg and Teff are undergoing thymocyte negative selection, the Teff cells are the source of IL-2, further emphasizing the interrelationship between cells from these two developmental pathways [70, 71]. The thymopoietic cytokine IL-7, which also induces pSTAT5, seems less important for tTreg development, especially since IL-7 production in the thymic medulla is not robust [72, 73]. The negative to low levels of expression of the CD127 subunit of the IL-7R by tTreg is consistent with these observations. Although experimentally established only in murine models, the importance of Teff-derived IL-2 for intrathymic tTreg development is likely to be true in humans as well.
An alternative developmental pathway comprised of a cd25- FoxP3low tTreg progenitor has also been described in mice. The conventional cd25+ and Foxp3low tTreg progenitors display distinct dynamics for the imprinting of the tTreg cell–specific epigenetic signatures and downstream gene expression [68, 69]. Both types of progenitors generate mature tTreg with equal efficiency, but with non-overlap** TCR repertoires, suggesting again the developmental importance of intrathymic TCR-mediated signaling [69]. A fraction of the Foxp3low tTreg progenitors were found in the thymic cortex as well as in the medulla, raising the interesting question of whether their TCR repertoire is shaped by reactivity with cortical antigens. Treg derived from the two separate tTreg progenitors acquire different antigen specificities and may therefore have different roles in immune regulation. tTreg derived from the cd25+ progenitors protect from experimental autoimmune encephalomyelitis while those derived from Foxp3low progenitors protect from colitis [69]. The relevance to human tTreg development of these provocative findings of two different intrathymic developmental pathways that generate functionally non-redundant tTreg populations in the periphery has not yet been established.
The thymic medulla in tTreg development and selection
The development of tTreg depends on the unique processes for TCR engagement during thymic development. The presentation of self-antigens to SP thymocytes occurs in the thymic medulla, and critically depends on medullary thymic epithelial cells (mTEC), which promiscuously express tissue-restricted antigens (TRA) otherwise specific to different peripheral tissues [74]. This process of promiscuous gene expression (PGE) has been aptly and vividly described as casting an immunological self shadow within the thymus to permit selection of the T cell repertoire [75]. As noted above, the intensity of the TCR signal determines whether medullary thymocytes adopt a fate of either a Teff or tTreg or, even undergo deletion, after exposure to TRA expressed by the mTEC.
The promiscuous expression in mTEC of thousands of genes encoding TRAs is driven by the transcription factor AIRE [75,76,77]. Adoptive transfer of T cells from Aire-deficient knockout mice into wild-type, T cell–depleted recipients resulted in autoimmunity [75]. However, unlike Foxp3 mutations, the T lymphocyte defect is not intrinsic to lymphoid progenitors, as hematopoietic progenitors from normal marrow donors transplanted into Aire-deficient recipients still resulted in autoimmunity, thus establishing that the defective thymic microenvironment, and the absence of Aire expression in mTEC, which underlies the autoimmune phenotype. The expression of TRA by Aire-expressing mTEC was required for intrathymic deletion of autoreactive conventional T cells with a high-affinity TCR for TRA-derived peptides. Although the repertoire of TRA collectively expressed by Aire+ mTEC is broad, individual mTEC only express a limited number of TRA at any given point in time [78, 79]. Single-cell analyses indicate intercellular variability and stochastic patterns of TRA expression [79, 80]. A stochastic mechanism makes it unlikely that the self-shadow cast by Aire+ mTEC remains comprehensive. More recent data from single-cell transcriptome profiling studies support observations that promiscuous gene expression of TRAs exhibits ordered co-expression, although the mechanisms underlying this instruction remain biologically indeterminate. Ordered co-expression and random spatial distribution of a diverse range of TRAs likely enhance their presentation and encounter with passing thymocytes, while maintaining mTEC identity [80].
The role of AIRE in control of autoimmunity was first established by its positional cloning as the molecular cause of a complex pathology observed in patients diagnosed with autoimmune polyglandular syndrome 1 (APS1; a.k.a. the autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) syndrome; discussed below). This causal link was subsequently confirmed in Aire knockout mice where disease penetrance was however shown to be dependent on other genetic influences [75,76,–77]. The AIRE gene, located on chromosome 21q22.3, encodes a protein with domains that mediate oligomerization with other proteins relevant for its function [79] and assist in nuclear sublocalization to super-enhancer regions of DNA [80,81,82]. Promiscuous expression of TRA is promoted by AIRE binding and inhibition of molecules that normally repress gene expression. These targets include a repressive complex of ATF7ip (activating transcription factor 7-interacting protein) and MBD1 (methyl CpG-binding protein), and hypomethylated lysine 4 of histone H3 (H3K4me0) [83,84,85,86,87,88]. The mechanism and structural basis for transactivation of TRA by AIRE are still under active investigation.
AIRE and the thymic medulla are also important for generation of Tregs during thymocyte selection [89,90,91,92]. The murine models indicate that T cell progenitors with intermediate affinity for the TRA expressed by Aire+ mTEC are selected as Treg, and Aire deficiency causes reduction in tTreg number [2, 9]. Aire expression may also redirect T cells from a conventional T cell to a Treg fate because identical TCR specificities were found both on T cells infiltrating autoimmune targets in Aire-deficient mice and Tregs in wild-type animals [9, 93]. Similarly, self-reactive TCR sequences expected to be confined to Treg cells in healthy subjects are instead found as part of the conventional T cell TCR repertoire in APS1 patients, indicating altered Treg differentiation as a consequence of AIRE deficiency [94]. Interactions and transfer of AIRE-dependent TRAs from mTEC to thymic dendritic cells (DC) are an additional and essential mechanism to secure tTreg differentiation and thus self-tolerance [95,96,97,98]. The necessity of antigen transfer from AIRE-expressing mTEC to DC emphasizes the likely importance of medullary architecture in TCR repertoire selection. Extrathymic AIRE-expressing cells (eTAC) have been found in peripheral lymphoid tissues of both mice and humans. These eTAC can delete autoreactive T cells in the periphery, possibly via the expression of a restricted repertoire of TRAs, but their ability to induce regulatory T cells has not been established [99, 100].
Not all TRA expression by mTEC is AIRE dependent. There is evidence in mice that another transcription factor, Fezf2, may induce Aire-independent promiscuous expression of TRA. Analyses of Fezf2-deficient mice demonstrate that different patterns of autoantibodies and autoimmune targets are detected when compared to Aire-deficient animals [101]. Because genes controlled by either Aire or Fezf2 account for approximately 60% of all TRA loci, other molecular mechanisms must likely be in play to regulate the promiscuous transcription of the remaining loci. However, these are not yet known. Recently, the Chd4 chromo helicase domain gene has been discovered as an upstream regulator of a smaller selection of both Aire- and Fezf2-controlled TRA loci [102]. Chd4-deficient mice develop autoimmunity with T lymphoid infiltration of peripheral targets, including the salivary gland, kidney, lung, and liver.
When something goes wrong: the emerging domain of monogenic autoimmunity
Since first proposed by Good, inborn errors of immunity can be viewed as “experiments of nature” which provide clear-cut information on the importance and function of certain genes [103]. In his formulation, these defects anticipated the knockout technologies that have powered genetic studies in laboratory mice and other experimental models. Recently, with the broadening of genome sequencing and engineering, discoveries of new disease causative genes and mutation-specific variants originally observed in humans and subsequently experimentally verified in mice have grown at an increased pace.
Primary defects of the lymphoid lineage in the generation of tTregs
According to the last published classification of primary immune deficiencies, there are 9 different gene defects that manifest with autoimmunity due to impaired Treg cells, defined as Tregopathies [104,105,106,107]. In addition to the classical immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome due to FOXP3 mutation, the best characterized are CD25, CTLA4, and LRBA deficiencies, that is the absence of molecules as highly relevant for tTreg development and function, as mentioned earlier in this review [108,109,110]. Other recognized Tregopathies relate to a number of other defects. Gain of function mutations of STAT3, the latent transcription factor downstream of many cytokine receptor families, including IL-10 and IL-23, impair FOXP3 and IL-17 regulation and consequently interfere with the Treg and Th17 balance [111]. Deficiencies in STAT5b expression result in Tregopathies as this transcription factor initiates the regulatory cascade of the IL-2-CD25-FOXP3 axis [111]. More recently, rare Tregopathies have been recognized to be caused by BACH2 mutations, a transcription factor that stabilizes FOXP3 expression and represses Teff differentiation. [112, 113]. BACH2 is highly expressed in resting tTreg to maintain their quiescence, and is downregulated by activation [114, 115]. Finally, defects in regular tTreg development and function have been observed in patients with mutations of the beta subunit of the IL-2 receptor (CD122) or DEF6, which is involved in CTLA4 membrane trafficking [116,117,118].
Here, we will describe in more detail the prototypic autoimmune diseases of tTreg associated with FOXP3, CD25, and CTLA4 mutations. Importantly, mutations in other genes affecting early stages of T cell development may manifest both profound immune deficiency with infections and autoimmune symptoms. Examples of such immune deficiencies are mutations of the recombination activating genes RAG1 and RAG2 necessary for V(D)J recombination and TCR repertoire formation, or of the Wiskott-Aldrich syndrome protein (WASP) resulting in defective antigen recognition in Wiskott-Aldrich syndrome. [107, 119,120,121]. The Omenn syndrome, which is caused by incomplete V(D)J recombination and abnormal TCR repertoire formation due to hypomorphic RAG1/2 mutations, constitutes an excellent example of impaired tolerance induction due to an oligoclonal TCR repertoire and the escape of autoreactive T cells from stringent negative selection [120]. In Omenn syndrome, the failure of thymocyte maturation results in abnormal crosstalk between the thymocytes, mTEC, and dendritic cells, and abnormal medullary architecture and function [120] Consequently, intrathymic selection is abnormal so self-reactive T cells are released and undergo clonal expansion in the periphery, inducing autoimmunity directed against the skin, GI tract, liver, and/or blood cells that resembles graft-versus-host disease. Importantly, altered TCR signaling strength in the presence of a normal capacity for V(D)J recombination also impairs Teff and tTreg differentiation and tolerance acquisition [122]. The increased genomic analyses of patients with autoimmunity are expected to lead to the discovery of new gene mutations that cause Treg abnormalities.
FOXP3 deficiency
Immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome is a rare but highly instructive example of a monogenic autoimmune disease of tTreg differentiation or function [106]. The analysis of a naturally occurring Foxp3 mutation in the scurfy mouse enabled several groups of investigators to identify IPEX to be the consequence of mutations in FOXP3, which is localized on the X chromosome [123]. The clinical phenotype of male scurfy mice matched the clinical presentation of 19 related males patients initially reported in 1982 by Powell and colleagues [124]. Subsequent studies in transgenic and genetically engineered mice established the link between FOXP3 mutations, a lack of tTreg, CD4+ T cell hyperproliferation, and tissue infiltration and destruction (reviewed in ref. 125). In most IPEX patients, the clinical features manifest themselves very early in life as severe multi-organ autoimmunity. In affected children, IPEX frequently presents in the first month of life and almost always within the first year [126, 127]. However, cases have been reported where the disease became apparent in utero and caused prenatal death, which highlights the importance of appropriate tTreg function early in life [128]. Enteropathy with watery refractory diarrhea is the typical common symptom of IPEX and is either preceded or followed in at least one-half of cases by autoimmune type 1 diabetes (T1D). Skin diseases in the form of severe eczema or psoriasis and cytopenias constitute other common features of the disease. As a result of both the dysfunctional tTreg suppressive activity and the intrinsic inability of mutated FOXP3 to regulate the cell cycle, CD4+ Teff cells are hyperproliferative [129]. Teff cell expansion is skewed towards a Th2 phenotype with more IL17 and IL22 production than in wild-type T cells, an imbalance that probably contributes to the development of autoimmunity. A secondary imbalance in the T follicular helper subsets and an increase in autoreactive B cells producing autoantibodies have also been described [130,131,132]. A subset of tTreg, T follicular regulatory (Tfr) cells, are normally found in germinal centers where they suppress T follicular helper (Tfh) cells and B cells to prevent autoantibody production [96, 133,134,135]. Although not yet established, autoantibody production in IPEX patients is likely due to Tfr defects, as shown in AIRE-deficient patients [96]. Despite the lack of functional FOXP3, CD4+ CD25+ CD127lo T cells are still present in IPEX patients. Phenotypic tTreg-like cells were also observed in mice carrying a null Foxp3 allele that was marked with GFP [136]. This murine model and the clinical findings indicate that expression of some phenotypic markers of tTreg is not FOXP3 dependent. However, FOXP3 mutations impair tTreg function, as measured by in vitro inhibition of proliferation and cytokine production by Teff [132].
In addition to the many preserved Treg phenotypic markers, the TSDR of the mutant FOXP3 locus in tTreg of IPEX patients is completely demethylated, as in healthy subjects’ tTreg. However, the frequency of tTreg quantified by TSDR demethylation in IPEX patients is higher than that of healthy subjects [131]. While the meaning of this observation remains to be clarified, the demethylation of the TSDR in IPEX indicates that some epigenetic modifications of the FOXP3 enhancer occur independently of FOXP3 expression during thymic Treg development, probably as a result of the intrathymic TCR or cytokine receptor–mediated signals. The fact that most relevant molecules involved in Treg identity and function have preserved expression in FOXP3-mutated Tregs, but that the cells remain dysfunctional, is an unexplained paradox that deserves further study. Consistent with the observation that FOXP3 activity is mediated by non-tTreg-specific transcription factors like TCF1, expression of Treg-associated genes can occur even when FOXP3 is mutated [33]. The FOXP3-mutated tTreg may contribute to the pathology in IPEX not only because they lack adequate suppressive activity, but also because their functional identity is unstable and they can acquire effector functions [137, 138]. This understanding is supported by enrichment of FOXP3+ T cells among the IL-17-producing cells and increased FOXP3+ CD161+ co-expressing cells in the IPEX gut mucosa (132 and unpublished observation).
Correlations between IPEX genotype and phenotype have not been consistent, and the reasons for this discrepancy remain so far unknown [127]. Unlike Foxp3 knockout mice, most patients are not null for FOXP3. Instead, expression is either decreased or mutant isoforms are generated that retain some biological activity. The incomplete loss of FOXP3 function probably explains the later onset of symptoms in patients compared to that of mice. As an example of the clinical heterogeneity, the clinical presentations and severity of IPEX can differ between siblings with identical FOXP3 mutations. While the heterogeneity could be explained by modifying genes, unknown epigenetic modifiers, or environmental triggers for autoimmunity, the relative contributions of these variables to the clinical phenotype of IPEX are not yet understood.
Treatment of IPEX patients can be particularly demanding because control of the severe autoimmunity often requires multiple immune suppressive drugs, in addition to supportive care for enteropathy or hormone replacement therapy, e.g., insulin for T1D. Of the different immune suppressive drugs, rapamycin is the most efficacious. Rapamycin suppresses Teff while sparing the Treg cells and can activate FOXP3-independent regulatory pathways to increase suppressive activity [139, 140]. The only curative therapy currently available to treat IPEX is allogeneic hematopoietic stem cell transplantation (HSCT), which replaces the patient’s immune system and allows for the normal development of donor-derived tTreg and Teff cells [126].
CD25 deficiency
Loss-of-function mutations of genes required for suppressive activity also cause tTreg dysfunction and autoimmunity. The first of these were mutations of CD25, the IL-2Rα subunit, in patients with an IPEX-like illness inherited in an autosomal recessive manner [108]. Like IPEX, patients with CD25 deficiency have severe, early-onset enteropathy and eczema. Other frequently observed autoimmune manifestations include alopecia, thyroiditis, and cytopenias. However, CD25 deficiency results in greater immune deficiency, especially susceptibility to severe viral infections, than is observed in IPEX.
The phenotype of CD25 deficiency is consistent with its role in formation of the high-affinity IL-2 receptor, which is essential for both tTreg and Teff functions. The mutations described so far in IPEX-like patients lead to abrogation of membrane CD25 expression [41, 108]. Consequently, both intrathymic tTreg progenitors and mature peripheral tTreg lose a major survival and differentiation signal. The rare tTreg that are detected still express FOXP3, CTLA4, and TIGIT, and have normal TSDR demethylation. Besides the reduced number, the CD25-tTreg are functionally abnormal because of loss of the critical IL-2 scavenging function, as well as decreased expression of IL-2-dependent genes [42]. Because the IL-2R is also required for Teff expansion in response to infectious stimuli, the loss of CD25 results in the immune deficiency and susceptibility to viral infection. The higher susceptibility for infections is due to impaired IL-2-dependent expansion of Teff cells needed to efficiently clear infections. The greater severity of the IPEX-like phenotype than the immune deficiency in the CD25-deficient patients suggests that there is less redundancy for CD25 in tTreg development and function than in Teff cells. Consistent with this hypothesis, polymorphisms in the CD25 locus that may decrease levels of CD25 expression are associated with susceptibility to T1D, with no evidence of immune deficiency [141]. The co-existence of autoimmunity and higher susceptibility for infections constitute a particular challenge in care of patients with CD25 deficiency. As for IPEX, the only available curative treatment for patients with CD25 deficiency is allogeneic HSCT [142].
CTLA4/LRBA deficiency
Expression of CTLA4 by tTreg is functionally important for the suppression of Teff via blocking and/or transendocytosis of the CD28 co-stimulatory receptor for CD80/CD86 expressed by APC. Thus, CTLA4 acts as a brake on Teff activation. High-level surface expression of CTLA4 depends on the Liposaccharide-responsive beige-like anchor (LRBA), a membrane-anchoring protein that promotes recirculation of membrane CTLA4 from cytoplasmic pools [143]. Heterozygosity for loss-of-function mutations of CTLA4 and biallelic LRBA mutations each result in impaired tTreg function and syndromic autoimmunity, either CTLA4 haploinsufficiency with autoimmune infiltration (CHAI) or LRBA deficiency with autoantibodies, Treg defects, autoimmune infiltration, and enteropathy (LATAIE), respectively [110, 144–146] . As in IPEX and CD25 deficiency, CHAI and LATAIE patients frequently present with enteropathy and cytopenias, along with and lymphadenopathy and hepatosplenomegaly due to lymphoproliferation. Pulmonary and neurological pathologies may also be observed. Some patients have also developed malignancies, including lymphoma, gastric cancer, and EBV-associated malignancies. A peculiarity of CTLA4 deficiency is the incomplete penetrance, which has been reported in a large study to be 67% [147]. There is also variance in disease severity within members of the same family carrying the identical mutation, rendering prognostication difficult. Both CHAI and LATAIE diseases can benefit from targeted treatment with abatacept, a recombinant fusion protein of CTLA4- and Ig Fc, which is a soluble molecule that can replace the function of membrane bound CTLA4 [110, 148]. Alternatively, mTOR inhibitors like rapamycin can inhibit CD28-mediated activation of Teff cells and provide benefit to these patients. LRBA-deficient cells can be induced in vitro to express CTLA4 after inhibition of lysosomal degradation, e.g., by chloroquine treatment, suggesting another potential avenue for pharmacologic treatment. However, HSCT remains the only potentially curative treatment, and is appropriate, in severe cases [147, 149].
Gene therapy to restore or repair the Treg subsets and re-establish immune regulation
The engineered replacement of defective genes has a great potential as a definitive treatment of PIRD due to tTreg defects. In contrast, pharmacologic immunomodulation, e.g., rapamycin to inhibit Teff, does not restore tTreg function and requires life-long treatment to control or prevent autoimmunity. HSCT from an allogeneic donor has high risks of treatment-related morbidity and mortality associated with the use of cytotoxic chemotherapy and graft-versus-host disease. Persistence of autoimmunity has been occasionally observed after HSCT, possibly because of survival of either autoimmune Teff, or slow or inadequate development of new donor-derived tTreg [126]. Preclinical testing in experimental in vitro and murine in vivo models have established both proof-of-concept and feasibility for gene therapy as a disease-specific approach to restore tTreg function in patients with inborn errors of FOXP3 or CD25. Targeting of the normal gene to HSC holds the promise of life-long production of genetically corrected blood cells. Because expression of genes like FOXP3, CD25, CTLA4, or LRBA is precisely controlled, successful gene therapy depends on the presence of each gene’s regulatory elements to assure appropriate cell type–specific expression, inductive conditions, timing, and levels of expression for the repaired sequence.
Two different gene therapy approaches have been explored to correct FOXP3 mutations in IPEX patients [129, 149, 150]. CRISPR/Cas9-based gene editing has been tested to insert a normal full-length FOXP3 coding sequence into mutant HSPC or T cells in vitro, using the endogenous regulatory elements to assure normal expression [149]. Lentiviral-mediated addition of the FOXP3 coding sequence, along with a cassette containing the putative regulatory elements, has also been shown to confer appropriate expression and restoration of tTreg functions [150]. Instead of targeting HSPC, lentiviral-mediated transfer of wild-type FOXP3 gene into pathogenic Teff can convert these cells into functional Treg. This lentiviral approach has been successful in murine models and is now being developed for a clinical trial [150]. For CD25 deficiency, an innovative virus-free CRISPR/Cas9 gene–editing approach has been shown to efficiently restore normal CD25 expression in mutant T cells [151]. Given the toxicities and incomplete normalization of immune function that have been observed with allogeneic HSCT, these novel gene therapy approaches offer a combination of decreased risk of treatment-related complications like GVHD and potential cure of the PIRD due to tTreg defects.
Impaired tTreg development due to abnormal thymic microenvironment
A number of primary genetic as well as acquired diseases of the thymic microenvironment result in immune dysregulation, marked by imbalances in thymic output between Teff and tTreg. The primary genetic diseases include those in which the function of an ostensibly normal thymic structure is impaired and others in which the formation of the thymic microenvironment is abnormal. Two examples of functional and developmental thymic defects are, respectively, APS1 due to AIRE mutation and the chromosome 22q11.2 deletion syndrome (22q11.2DS).
Autoimmune polyglandular syndrome 1
The AIRE gene and pathogenic mutations were identified by positional cloning of the gene responsible for APS1, which was originally called autoimmune polyendocrinopathy candidiasis ectodermal dysplasia (APECED) [76, 77]. As discussed elsewhere in this issue, the classical manifestations of APS1 were a triad of chronic mucocutaneous candidiasis (CMC), hypoparathyroidism, and adrenal insufficiency, but the clinical phenotype is expanding with improved detection of AIRE mutations. This is analogous to the situation with IPEX where broader availability of genetic testing for FOXP3 mutations has led to an expanded range of clinical manifestations and disease progression than predicted with the classical definition. An intriguing difference observed between APS1 and the engineered mouse model of Aire deficiency is the observation that patients present with decreased peripheral numbers of tTreg, which is not seen in the Aire− mice, despite their tTreg developmental defect. In addition to the altered TCR repertoire of tTreg due to intrathymic defects in selection, the tTreg from APS1 patients have intrinsic defects of decreased FOXP3 expression and function [89].
Given the dual role of AIRE expression by mTEC in clonal deletion of autoreactive conventional T cells and the positive selection of tTreg or re-direction of some cells to tTreg differentiation, the relative contributions of each defect in APS1 is an open question. For example, if the aberrant negative selection was accompanied by normal tTreg generation, would autoimmunity still occur? In athymic Nude mice transplanted with both a normal and Aire-deficient thymus, autoimmunity still developed despite the presumed normal generation of Treg by one of the thymi [152]. It is not known if the relative tTreg output of the mouse thymus is the same as human, nor is it known if transplanted thymus is equally as efficient as a native organ. Nevertheless, if these murine results were also true of APS1 patients, they would have significant therapeutic implications. First, if there were a therapy that allowed normal tTreg generation (such as the transplantation of fresh or cultured tissue [153] or in the future a functionally competent yet artificial thymic microenvironment), then thymectomy to remove the native AIRE-deficient thymus would be necessary to prevent further production of autoreactive Teff cells. Second, the efficacy of the newly produced tTreg would likely be enhanced if ablative lymphocytotoxic therapy was first administered to eliminate autoreactive T cells that had already been made. Anticipated risks of immunoablation include infection susceptibility such as occurs after HSCT, or transient autoimmunity due to imbalances of homeostatic expansion of Teff and tTreg populations in the lymphopenic environment.
The discovery of other genes and pathways like Fezf2 and Chd4 that regulate mTEC development and TRA expression in murine models make it likely that other defects of intrathymic production of tTreg will be discovered in patients with autoimmunity or other forms of immune dysregulation [101, 102]. To date, a clinical manifestation of human FEZF2 mutation has not yet been described. Since Fezf2 deletion is lethal in knockout mice because of brain and behavioral abnormalities, deletion or complete loss-of-function mutants are likely to be nonviable, but hypomorphic variants that specifically interfere with mTEC functions might be found in patients with immune dysregulatory disorders.
COPA syndrome
In 2015, a cohort of five families with an autosomal dominant syndrome of autoantibodies, arthritis, and interstitial lung disease were identified by whole exome sequencing as having predicted loss-of-function mutations in the Coatamer protein complex alpha (COPA) subunit gene [154]. COPA is required for transport of proteins from the endoplasmic reticulum (ER) to the Golgi apparatus as well as retrograde transport of selected proteins from the Golgi back to the ER like those with dilysine tags. The mutations in the COPA gene involve a WD motif that result in decreased binding to dilysine motifs and an impairment of intracellular protein trafficking. The patients with the COPA syndrome have decreased frequencies of interferon gamma–secreting Th1 cells and increased IL-17A-secreting Th17 cells. The reported autoantibodies have been classified as antinuclear, anti-neutrophil (ANCA), and rheumatoid factor-like, but unlike APS1, anti-interferon or other anti-cytokine antibodies have not been observed [154, 155]. Mice with a knock-in of one of the mutations observed in the original kindreds phenocopied clinical manifestations like interstitial and lung disease, as well as T cell activation and dysfunction [156]. The mechanism of autoimmunity in the COPA-deficient mice is the defective selection of autoreactive conventional T cells and decreased production of Treg. One hypothesis to explain this abnormal selection is inadequate expression of self-antigens, hence a mechanism analogous to that operational in APS1. However, COPA-deficient mice also show an abnormal expression of stimulator of interferon genes (STING), suggesting that there may be an excess in interferon-related inflammation [157]. The effects of aberrant intrathymic inflammatory signaling on tTreg generation are as yet unknown. The COPA syndrome raises the exciting possibility that other monogenic causes of specific autoimmunity will be found. For APS1, COPA, and any other autoimmune syndromes due to abnormal selection of conventional Teff cells and Tregs, key questions remain about the nature of TRA expression by the respective mTEC, the autoantigens recognized by the peripheral T cell populations, and the resultant autoimmune manifestations.
Defects of thymic ontogeny leading to abnormal tTreg production
There are human diseases, notably 22q11.2DS and CHARGE syndrome, discussed elsewhere in this issue, which cause failure of thymic organogenesis, T lymphopenia, and increased risk of autoimmunity [10, 158,158,, 159]. The 22q11.2DS syndrome results from interstitial deletion of 1.5–3.0 Mb of DNA on one copy of the long arm of chromosome 22 encompassing approximately 30–100 genes, of which hemizygosity for two genes, TBX1 and CRKL, causes thymic hypoplasia in murine models [160]. The majority of patients with non-SCID levels of T lymphopenia are at risk not only for opportunistic infection but also high risk of autoimmune or allergic diseases [10, 161, 162]. The most commonly observed forms of autoimmunity are juvenile rheumatoid arthritis, autoimmune cytopenias, and thyroiditis. There is also a high incidence of allergy in the form of atopic skin diseases, asthma, and food allergies [163]. Analyses of the peripheral tTreg populations in 22q11.2DS patients have demonstrated overall reduction in the number of phenotypic CD4+ CD25hi CD127low FOXP3+ Treg, but their suppressive function is preserved [164] and unpublished observation). However, another study found abnormal Treg function in both intrathymic Treg progenitors and peripheral Treg populations [165]. Transplantation of allogeneic normal thymic tissue has been shown to restore peripheral Treg numbers, consistent with the Treg defects in 22q11.2DS patients being secondary to the stromal defect [153, 166].
Analyses of the thymus in patients with 22q11.2 DS has shown smaller than normal-sized medullary regions, reduced numbers of mTEC, and crucially decreased numbers of AIRE+ mTEC [165]. The lack of AIRE(+) cells is compatible with blocks in differentiation that prevent the maturation of AIRE− mTEC into later AIRE+ stages. The overall structure of the medullary regions is disorganized, which has been compared to an immature thymus. While a differentiation block is an attractive explanation for the thymic pathologies observed in the 22q11.2DS, the tools for topological analyses are not well enough advanced to distinguish “immaturity” from pathological disorder, e.g., an abnormal structure due to aberrant gene expression that are not simply a block of mTEC differentiation. The selection of tTreg progenitors requires topologically complex interactions with mTEC and DC, so pathological disorganization of the thymic medulla in 22q11.2DS is likely to be at least one cause of the abnormal tTreg production.
Besides the thymic hypoplasia, hemizygosity for DGCR8 in 22q11.2DS provides a link to the autoimmunity of APS1. AIRE regulates a specific set of microRNAs, which are aberrantly expressed in AIRE− murine and human mTEC [167]. Some of these microRNA regulate promiscuous gene expression, with miR29a being a specific regulator of AIRE-dependent genes. DGCR8 is a component of the microprocessor complex, which cleaves canonical miRNAs. Mice with TEC-specific deletion of DGCR8 develop thymic hypoplasia and decreased expression of Aire in their mTEC [168]. The hemizygosity for DGCR8 in the 22q11.2DS thymus is not expected to result in the same profound loss as in the homozygous Dgcr8 murine knockouts, but the expected decrease in DGCR8 expression and possibly microprocessor complex activity could contribute to defective AIRE expression and tTreg development.
Relevance to acquired diseases of tTreg generation
Thymic dysfunction and/or hypoplasia associated with decreased Treg numbers and autoimmunity has also been noted for acquired pathologies. Approximately 45% of patients with thymic epithelial tumors (TET, “thymoma”) have evidence of autoimmunity. While initially thought to be restricted to myasthenia gravis, extensive characterization of TET patients has shown endocrine disorders, chronic mucocutaneous candidiasis (CMC), and cytopenias like those seen in APS1 [169]. A proposed mechanism is the ability of these tumors to support T cell development but an inability to either delete autoreactive cells or generate normal tTreg because of a lack of expression of AIRE in the tumors [170, 171]. In support of this hypothesis, the clinical autoimmune presentations and range of autoantigens detected in TET patients resembles APS1 [172]. However, these patients frequently develop myasthenia gravis with autoantibodies directed against the acetylcholine receptor, a phenotype rarely seen in APS1. There is both an overlap and differences in autoantibodies directed against interferons in APS-1 (interferon ω) and TET patients with myasthenia (interferon α’s), suggesting that there are subtle differences in the patterns of autoreactivity accounting for phenotypic differences [173]. The general hypothesis of perturbed T cell development could be extended to include lack of expression by the tumor cells of other thymic medullary genes required to properly shape the T cell repertoire, e.g., COPA or possibly FEZF2 or CHD4, which were discussed above. These tumors have not yet been comprehensively compared to normal thymic tissue, and such analyses could reveal non-AIRE-dependent mechanisms that contribute to failure to delete autoreactive, conventional T cells or to promote the development of tTreg. Complicating the interpretation of the peripheral T cells is the co-existence of both TET and normal thymus tissue. The development of autoimmunity may reflect the inability of the normal thymic tissue to produce sufficient tTreg to compensate for the autoreactive conventional T cells aberrantly generated within the tumor. Surgical removal of the tumor frequently results in amelioration of the autoimmune symptoms, but many patients have persistent autoimmunity and may even require immunosuppressive therapy.
Another relevant condition in which thymic dysfunction is likely to contribute to abnormal T cell repertoire formation and autoimmunity is chronic graft-versus-host disease (cGVHD) occurring after allogeneic hematopoietic stem cell transplantation [174]. Acute GVHD (aGVHD) occurs when histoincompatible T lymphocytes engraft in an immunosuppressed transplant recipient, usually resulting in T cell–mediated inflammatory injury to targets of the skin, gastrointestinal tract, and/or liver. In contrast to aGVHD, there is fibrosis and frequent incidence of autoimmune diseases in cGVHD. The relationship between acute and chronic GVHD is complex: different pathologies (inflammation vs fibrosis, not all patients with aGVHD develop cGVHD, cGVHD can develop in patients with no prior evidence of clinical aGVHD, and alloreactivity vs autoimmunity). Patients with aGVHD have decreased levels of circulating Treg, while paradoxically, cGVHD patients have higher numbers of circulating Treg [175]. It has been long known that the thymic epithelium is injured in aGVHD, but the clinical implications difficult to study in humans [176, 177]. A pathophysiologic linkage between aGVHD and cGVHD has been provided by analyses of the immunologic effects of TEC damage caused by aGVHD in murine transplant recipients. TEC damage by alloreactive T cells results in loss of Aire+ mTEC and prevents the deletion of autoreactive cells [178, 179]. The effects on Treg populations were not studied, but based on the dual roles of the mTEC in deletion of autoreactive conventional thymocytes and promotion of tTreg development, it is reasonable to speculate that tTreg production is also impaired. Overall, the data from GVHD experiments indicate that analogously with APS1 or 22q11.2DS, secondary defects of mTEC number and function leading to abnormal tTreg generation are relevant to the acquired autoimmunity observed in HSCT recipients.
Conclusions
Just as monogenic diseases causing clinical immune deficiency provided a window into how the immune system controls infection, the primary immune regulatory disorders are illuminating the mechanism by which autoimmunity is prevented. Among those, the diseases that interfere with tTreg development or function have established the importance of transcription factors like FOXP3 and AIRE, and the roles of the differentiation program of tTreg progenitors and the thymic medulla in tTreg development. The clinical entities of IPEX and APS1 caused by the mutations of FOXP3 and AIRE, respectively, are likely to be the first of a string of new primary monogenic diseases that can be used for discovery of immune regulatory mechanisms and targeted interventions.
References
Plitas G, Rudensky AY (2016) Regulatory T cells: differentiation and function. Cancer Immunol Res 4:721–725
Owen DL, Sjaastad LE, Farrar MA (2019) Regulatory T cell development in the thymus. J Immunol 203:2031–2041
Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N (2020) Regulatory T cells and human disease. Annu Rev Immunol 38:541–566
Ramsdell F, Rudensky AY (2020) Foxp3: a genetic foundation for regulatory T cell differentiation and function. Nat Immunol 21:708–709
Tran DQ, Ramsey H, Shevach EM (2007) Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 110:2983–2990
Georgiev P, Charbonnier LM, Chatila TA (2019) Regulatory T cells: the many faces of Foxp3. J Clin Immunol 39:623–640
Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N (2018) The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity 49:1004–1019
Shevach EM (2000) Suppressor T cells: rebirth, function and homeostasis. Curr Biol 10:R572–R575
Savage PA, Klawon DEJ, Miller CH (2020) Regulatory T cell development. Annu Rev Immunol 38:421–453
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS (2015) 22q11.2 deletion syndrome. Nat Rev Dis Primers 1:15071
Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, Shultz LD (2017) Humanized mouse models of clinical disease. Annu Rev Pathol 12:187–215
Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, Zhu Y, Kim K, Kohn DB, Baltimore D, Crooks GM, Montel-Hagen A (2017) Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods 14:521–530
Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, Sacchetti N, Kawai T, Garabedian EK, Draper D, Bergerson JRE, DeRavin SS, Freeman AF, Gungor T, Hartog N, Holland SM, Kohn DB, Malech HL, Markert ML, Weinacht KG, Villa A, Seet CS, Montel-Hagen A, Crooks GM, Notarangelo LD (2020) Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv 4:2611–2616
MacDonald KN, Piret JM, Levings MK (2019) Methods to manufacture regulatory T cells for cell therapy. Clin Exp Immunol 197:52–63
Raffin C, Vo LT, Bluestone JA (2020) Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol 20:158–172
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH (2001) Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med 193:1285–1294
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA (2001) CD4+CD25high regulatory cells in human peripheral blood. J Immunol 167:1245–1253
Levings MK, Sangregorio R, Roncarolo MG (2001) Human CD25(+)CD4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med 193:1295–1302
Barzaghi F, Passerini L, Gambineri E, Ciullini Mannurita S, Cornu T, Kang ES, Choe YH, Cancrini C, Corrente S, Ciccocioppo R, Cecconi M, Zuin G, Discepolo V, Sartirana C, Schmidtko J, Ikinciogullari A, Ambrosi A, Roncarolo MG, Olek S, Bacchetta R (2012) Demethylation analysis of the FOXP3 locus shows quantitative defects of regulatory T cells in IPEX-like syndrome. J Autoimmun 38:49–58
Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, McCune JM (2010) Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 330:1695–1699
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St Groth B, Clayberger C, Soper DM, Ziegler SF, Bluestone JA (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203:1701–1711
Hori S (2014) Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev 259:159–172
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27:68–73
Lu L, Barbi J, Pan F (2017) The regulation of immune tolerance by FOXP3. Nat Rev Immunol 17:703–717
Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, Ziegler SF, Roncarolo MG, Levings MK (2005) The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest 115:3276–3284
Smith EL, Finney HM, Nesbitt AM, Ramsdell F, Robinson MK (2006) Splice variants of human FOXP3 are functional inhibitors of human CD4+ T-cell activation. Immunology 119:203–211
Joly AL, Seitz C, Liu S, Kuznetsov NV, Gertow K, Westerberg LS, Paulsson-Berne G, Hansson GK, Andersson J (2018) Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ Res 122:1385–1394
Frith K, Joly AL, Ma CS, Tangye SG, Lohse Z, Seitz C, Verge CF, Andersson J, Gray P (2019) The FOXP3Δ2 isoform supports Treg cell development and protects against severe IPEX syndrome. J Allergy Clin Immunol 144:317–320.e8
Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J (2015) Foxp3 lacking exons 2 and 7 is unable to confer suppressive ability to regulatory T cells in vivo. J Autoimmun 63:23–30
Mailer RKW (2018) Alternative splicing of FOXP3-virtue and vice. Front Immunol 9:530
Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY (2007) Genome-wide analysis of Foxp3 target genes in develo** and mature regulatory T cells. Nature 445:936–940
van der Veeken J, Glasner A, Zhong Y, Hu W, Wang Z-M, Bou-Puerto R, Charbonnier L-M, Chatila TA, Leslie CS, Rudensky AY (2020) The transcription factor Foxp3 shapes regulatory T cell identity by tuning the activity of trans-acting intermediaries. Immunity 53:971–984
Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, **asena D, Sharma SM, McCadden EM, Getnet D, Drake CG, Liu JO, Ostrowski MC, Pardoll DM (2009) Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science 325:1142–1146
Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat Rev Immunol 8:523–532
Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA (2004) Role of LAG-3 in regulatory T cells. Immunity 21:503–513
Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA (2011) Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev 241:180–205
Mellor AL, Munn DH (2004) IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 4:762–774
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ (2007) Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27:635–646
O’Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK (2009) The initial phase of an immune response functions to activate regulatory T cells. J Immunol 183:332–339
Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, Doglioni C, Ponzoni M, Cicalese MP, Assanelli A, Tommasini A, Brigida I, Dellepiane RM, Martino S, Olek S, Aiuti A, Ciceri F, Roncarolo MG, Bacchetta R (2013) Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol 146:248–261
Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY (2016) An essential role for the IL-2 receptor in Treg cell function. Nat Immunol 17:1322–1333
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265
Fan X, Rudensky AY (2016) Hallmarks of tissue-resident lymphocytes. Cell 164:1198–1211
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, Nomura T, Ono M, Amoura Z, Gorochov G, Sakaguchi S (2009) Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30:899–911
Barbi J, Pardoll D, Pan F (2014) Treg functional stability and its responsiveness to the microenvironment. Immunol Rev 259:115–139
Shevyrev D, Tereshchenko V (2019) Treg heterogeneity, function, and homeostasis. Front Immunol 10:3100
Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C (2018) Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol 19:291–301
Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO (2010) Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol 11:618–627
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immu-noregulatory T cells that control autoimmune diabetes. Immunity 12:431–440
Newton R, Priyadharshini B, Turka LA (2016) Immunometabolism of regulatory T cells. Nat Immunol 17:618–625
Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J (2007) Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 5:e38
Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, Turbachova I, Hamann A, Olek S, Huehn J (2007) DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 37:2378–2389
Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, Lio CW, Li X, Huang Y, Vijayanand P, Lähdesmäki H, Rao A (2016) Control of Foxp3 stability through modulation of TET activity. J Exp Med 213:377–397
Nair VS, Song MH, Ko M, Oh KI (2016) DNA demethylation of the Foxp3 enhancer is maintained through modulation of ten-eleven-translocation and DNA methyltransferases. Mol Cell 39:888–897
Nair VS, Oh KI (2014) Down-regulation of Tet2 prevents TSDR demethylation in IL2 deficient regulatory T cells. Biochem Biophys Res Commun 450:918–924
Baron U, Werner J, Schildknecht K, Schulze JJ, Mulu A, Liebert UG, Sack U, Speckmann C, Gossen M, Wong RJ, Stevenson DK, Babel N, Schurmann D, Baldinger T, Bacchetta R, Grutzkau A, Borte S, Olek S (2018) Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci Transl Med 10:eaan3508
Lio CW, Hsieh CS (2008) A two-step process for thymic regulatory T cell development. Immunity. 28:100–111
Li MO, Rudensky AY (2016) T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16:220–233
Hsieh CS, Lee HM, Lio CW (2012) Selection of regulatory T cells in the thymus. Nat Rev Immunol 12:157–167
Breed ER, Watanabe M, Hogquist KA (2019) Measuring thymic clonal deletion at the population level. J Immunol 202:3226–3233
Vanhanen R, Leskinen K, Mattila IP, Saavalainen P, Arstila TP (2020) Epigenetic and transcriptional analysis supports human regulatory T cell commitment at the CD4+CD8+ thymocyte stage. Cell Immunol 347:104026
Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY (2006) An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol 7:401–410
Lee HM, Bautista JL, Scott-Browne J, Mohan JF, Hsieh CS (2012) A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity 37:475–486
Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW (2004) Autoreactive T cells in healthy individuals. J Immunol 172:5967–5972
Su LF, Del Alcazar D, Stelekati E, Wherry EJ, Davis MM (2016) Antigen exposure shapes the ratio between antigen-specific Tregs and conventional T cells in human peripheral blood. Proc Natl Acad Sci U S A 113:E6192–E61E8
Santoni de Sio FR, Passerini L, Restelli S, Valente MM, Pramov A, Maccari ME, Sanvito F, Roncarolo MG, Porteus M, Bacchetta R (2018) Role of human forkhead box P3 in early thymic maturation and peripheral T-cell homeostasis. J Allergy Clin Immunol 142:1909–21 e9
Herppich S, Toker A, Pietzsch B, Kitagawa Y, Ohkura N, Miyao T, Floess S, Hori S, Sakaguchi S, Huehn J (2019) Dynamic Imprinting of the Treg cell-specific epigenetic signature in develo** thymic regulatory T cells. Front Immunol 10:2382
Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, Ruscher R, Huang W, Proekt I, Miller CN, Hekim C, Jeschke JC, Aggarwal P, Broeckel U, LaRue RS, Henzler CM, Alegre ML, Anderson MS, August A, Marson A, Zheng Y, Williams CB, Farrar MA (2019) Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol 20:195–205
Owen DL, Mahmud SA, Vang KB, Kelly RM, Blazar BR, Smith KA, Farrar MA (2018) Identification of cellular sources of IL-2 needed for regulatory T cell development and homeostasis. J Immunol 200:3926–3933
Hemmers S, Schizas M, Azizi E, Dikiy S, Zhong Y, Feng Y, Altan-Bonnet G, Rudensky AY (2019) IL-2 production by self-reactive CD4 thymocytes scales regulatory T cell generation in the thymus. J Exp Med 216:2466–2478
Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA (2008) IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol 181:3285–3290
Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, Farrar MA (2008) Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 28:112–121
Derbinski J, Schulte A, Kyewski B, Klein L (2001) Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol 2:1032–1039
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D (2002) Projection of an immunological self shadow within the thymus by the aire protein. Science 298:1395–1401
Finnish-German AC (1997) An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 17:399–403
Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N (1997) Positional cloning of the APECED gene. Nat Genet 17:393–398
Derbinski J, Pinto S, Rosch S, Hexel K, Kyewski B (2008) Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc Natl Acad Sci U S A 105:657–662
Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, Holländer GA (2014) Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res 24:1918–1931
Dhalla F, Baran-Gale J, Maio S, Lia Chappell L, Holländer GA, Ponting CP (2020) Biologically indeterminate yet ordered promiscuous gene expression in single medullary thymic epithelial cells. EMBO J 39:e101828
Abramson A, Giraud M, Benoist C, Diane MD (2010) Aire’s partners in the molecular control of immunological tolerance. Cell 140:123–135
Pitkänen J, Vähämurto P, Krohn K, Peterson P (2001) Subcellular localization of the autoimmune regulator protein. characterization of nuclear targeting and transcriptional activation domain. J Biol Chem 276:19597–19602
Kumar PG, Laloraya M, Wang CY, Ruan QG, Davoodi-Semiromi A, Kao KJ, She JX (2001) The autoimmune regulator (AIRE) is a DNA-binding protein. J Biol Chem 276:41357–41364
Org T, Chignola F, Hetenyi C, Gaetani M, Rebane A, Liiv I, Maran U, Mollica L, Bottomley MJ, Musco G, Peterson P (2008) The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep 9:370–376
Koh AS, Kingston RE, Benoist C, Mathis D (2010) Global relevance of Aire binding to hypomethylated lysine-4 of histone-3. Proc Natl Acad Sci U S A 107:13016–13021
Waterfield M, Khan IS, Cortez JT, Fan U, Metzger T, Greer A, Fasano K, Martinez-Llordella M, Pollack JL, Erle DJ, Su M, Anderson MS (2014) The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat Immunol 15:258–265
Bansal K, Yoshida H, Benoist C, Mathis D (2017) The transcriptional regulator Aire binds to and activates super-enhancers. Nat Immunol 18:263–273
Huoh YS, Wu B, Park S, Yang D, Bansal K, Greenwald E, Wong WP, Mathis D, Hur S (2020) Dual functions of Aire CARD multimerization in the transcriptional regulation of T cell tolerance. Nat Commun 11:1625
Kekalainen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pontynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP (2007) A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol 178:1208–1215
Laakso SM, Laurinolli TT, Rossi LH, Lehtoviita A, Sairanen H, Perheentupa J, Kekalainen E, Arstila TP (2010) Regulatory T cell defect in APECED patients is associated with loss of naive FOXP3(+) precursors and impaired activated population. J Autoimmun 35:351–357
Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND, Savage PA (2013) Aire-dependent thymic development of tumor-associated regulatory T cells. Science 339:1219–1224
Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D (2015) Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348:589–594
Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA (2016) Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 44:1102–1113
Perry JSA, Lio CJ, Kau AL, Nutsch K, Yang Z, Gordon JI, Murphy KM, Hsieh CS (2014) Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity 41:414–426
Gallegos AM, Bevan MJ (2004) Central tolerance to tissue-specific antigens mediated by direct and indirect antigen presentation. J Exp Med 200:1039–1049
Sng J, Ayoglu B, Chen JW, Schickel J-N, Ferre EMN, Glauzy S, Romberg N, Hoenig M, Cunningham-Rundles C, Utz PJ, Lionakis MS, Meffre E (2019) AIRE expression controls the peripheral selection of autoreactive B cells. Sci Immunol 4(34):eaav6778
Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, Kline DE, Kline J, Vander Griend DJ, Huang H, Socci ND, Savage PA (2016) Dendritic cells coordinate the development and homeostasis of organ-specific regulatory T cells Immunity 44:847–859
Perry JSA, Russler-Germain EV, Zhou YW, Purtha W, Cooper ML, Choi J, Schroeder MA, Salazar V, Egawa T, Lee BC, Abumrad NA, Kim BS, Anderson MS, DiPersio JF, Hsieh CS (2018) Transfer of cell-surface antigens by scavenger receptor CD36 promotes thymic regulatory T cell receptor repertoire development and allo-tolerance. Immunity 48:923–36 e4
Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, Johannes KP, Su MA, Chang HY, Krummel MF, Anderson MS (2008) Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321:843–847
Gardner JM, Metzger TC, McMahon EJ, Au-Yeung BB, Krawisz AK, Lu W, Price JD, Johannes KP, Satpathy AT, Murphy KM, Tarbell KV, Weiss A, Anderson MS (2013) Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4(+) T cells Immunity 39:560–572
Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, Kodama T, Takayanagi H (2015) Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell 163:975–987
Tomofuji Y, Takaba H, Suzuki HI, Benlaribi R, Martinez CDP, Abe Y, Morishita Y, Okamura T, Taguchi A, Kodama T, Takayanagi H (2020) Chd4 choreographs self-antigen expression for central immune tolerance. Nat Immunol 21:892–901
Good RA (1954) Agammaglobulinemia: a provocative experiment of nature. Bull Univ Minn Hosp 26:1–19
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova JL, Sullivan KE (2020) Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 40:24–64
Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Puck J, Torgerson TR, Casanova JL, Sullivan KE, Tangye SG (2020) Human inborn errors of immunity: 2019 update of the IUIS Phenotypical Classification. J Clin Immunol 40:66–81
Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG (2018) Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 142:1679–1695
Notarangelo LD, Bacchetta R, Casanova JL, Su HC (2020) Human inborn errors of immunity: an expanding universe. Sci Immunol 5:eabb1662
Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW (2007) CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol 119:482–487
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, Frucht DM, Dumitriu B, Scheinberg P, Folio LR, Frein CA, Price S, Koh C, Heller T, Seroogy CM, Huttenlocher A, Rao VK, Su HC, Kleiner D, Notarangelo LD, Rampertaap Y, Olivier KN, McElwee J, Hughes J, Pittaluga S, Oliveira JB, Meffre E, Fleisher TA, Holland SM, Lenardo MJ, Tangye SG, Uzel G (2014) Immune dysregulation in human subjects with heterozygous germline mu- tations in CTLA4. Science 345:1623–1627
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB (2015) Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349:436–440
Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppänen M, Lernmark Å, Mustjoki S, Otonkoski T, Kere J, Morgan NG, Ellard S, Hattersley AT (2014) Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet 46:812–814
Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB (2006) Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol 177:2770–2774
Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, Sciumè G, Zare H, Vahedi G, Dema B, Yu Z, Liu H, Takahashi H, Rao M, Muranski P, Crompton JG, Punkosdy G, Bedognetti D, Wang E, Hoffmann V, Rivera J, Marincola FM, Nakamura A, Sartorelli V, Kanno Y, Gattinoni L, Muto A, Igarashi K, O’Shea JJ, Restifo NP (2013) BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature 498:506–510
Afzali B, Grönholm J, Vandrovcova J, O’Brien C, Sun HW, Vanderleyden I, Davis FP, Khoder A, Zhang Y, Hegazy AN, Villarino AV, Palmer IW, Kaufman J, Watts NR, Kazemian M, Kamenyeva O, Keith J, Sayed A, Kasperaviciute D, Mueller M, Hughes JD, Fuss IJ, Sadiyah MF, Montgomery-Recht K, McElwee J, Restifo NP, Strober W, Linterman MA, Wingfield PT, Uhlig HH, Roychoudhuri R, Aitman TJ, Kelleher P, Lenardo MJ, O’Shea JJ, Cooper N, Laurence ADJ (2017) BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol 18:813–823
Grant FM, Yang J, Nasrallah R, Clarke J, Sadiyah F, Whiteside SK, Imianowski CJ, Kuo P, Vardaka P, Todorov T, Zandhuis N, Patrascan I, Tough DF, Kometani K, Eil R, Kurosaki T, Okkenhaug K, Roychoudhuri R (2020) BACH2 drives quiescence and maintenance of resting Treg cells to promote homeostasis and cancer immunosuppression. J Exp Med 217:e20190711
Fernandez IZ, Baxter RM, Garcia-Perez JE, Vendrame E, Ranganath T, Kong DS, Lundquist K, Nguyen T, Ogolla S, Black J, Galambos C, Gumbart JC, Dawany N, Kelsen JR, de Zoeten EF, Quinones R, Eissa H, Verneris MR, Sullivan KE, Rochford R, Blish CA, Kedl RM, Dutmer CM, Hsieh EWY (2019) A novel human IL2RB mutation results in T and NK cell-driven immune dysregulation. J Exp Med 216:1255–1267 Correction: a novel human IL2RB mutation results in T and NK cell-driven immune dysregulation. 2019. J Exp Med 216:1465
Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, Modis Y, Alazami AM, Acres M, Haller W, Bowen C, Döffinger R, Sinclair J, Brothers S, Zhang Y, Matthews HF, Naudion S, Pelluard F, Alajlan H, Yamazaki Y, Notarangelo LD, Thaventhiran JE, Engelhardt KR, Al-Mousa H, Hambleton S, Rooryck C, Smith KGC, Lenardo MJ (2019) Human interleukin-2 receptor beta mutations associated with defects in immunity and peripheral tolerance. J Exp Med 216:1311–1327
Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N, Meeths M, Krolo A, Yüce Petronczki Ö, Pfajfer L, Hou TZ, Halliday N, Santos-Valente E, Kalinichenko A, Kennedy A, Mace EM, Mukherjee M, Tesi B, Schrempf A, Pickl WF, Loizou JI, Kain R, Bidmon-Fliegenschnee B, Schickel JN, Glauzy S, Huemer J, Garncarz W, Salzer E, Pierides I, Bilic I, Thiel J, Priftakis P, Banerjee PP, Förster-Waldl E, Medgyesi D, Huber WD, Orange JS, Meffre E, Sansom DM, Bryceson YT, Altman A, Boztug K (2019) Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis. Nat Commun 10:3106
Notarangelo LD, Kim MS, Walter JE, Lee YN (2016) Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 16:234–246
Villa A, Notarangelo LD (2019) RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev 287:73–90
Candotti F (2018) Clinical manifestations and pathophysiological mechanisms of the Wiskott-Aldrich Syndrome. J Clin Immunol 38:13–27
Rowe JH, Delmonte OM, Keles S, Stadinski BD, Dobbs AK, Henderson LA, Yamazaki Y, Allende LM, Bonilla FA, Gonzalez-Granado LI, Celikbilek Celik S, Guner SN, Kapakli H, Yee C, Pai SY, Huseby ES, Reisli I, Regueiro JR, Notarangelo LD (2018) Patients with CD3G mutations reveal a role for human CD3gamma in Treg diversity and suppressive function. Blood 131:2335–2344
Godfrey VL, Wilkinson JE, Russell LB (1991) X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol 138:1379–1387
Powell BR, Buist NRM, Stenzel P (1982) An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr 100:731–737
Bacchetta R, Barzaghi F, Roncarolo MG (2018) From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci 1417:5–22
Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, Nademi Z, Slatter MA, Ulloa ER, Shcherbina A, Roppelt A, Worth A, Silva J, Aiuti A, Murguia-Favela L, Speckmann C, Carneiro-Sampaio M, Fernandes JF, Baris S, Ozen A, Karakoc-Aydiner E, Kiykim A, Schulz A, Steinmann S, Notarangelo LD, Gambineri E, Lionetti P, Shearer WT, Forbes LR, Martinez C, Moshous D, Blanche S, Fisher A, Ruemmele FM, Tissandier C, Ouachee-Chardin M, Rieux-Laucat F, Cavazzana M, Qasim W, Lucarelli B, Albert MH, Kobayashi I, Alonso L, Diaz De Heredia C, Kanegane H, Lawitschka A, Seo JJ, Gonzalez-Vicent M, Diaz MA, Goyal RK, Sauer MG, Yesilipek A, Kim M, Yilmaz-Demirdag Y, Bhatia M, Khlevner J, Richmond Padilla EJ, Martino S, Montin D, Neth O, Molinos-Quintana A, Valverde-Fernandez J, Broides A, Pinsk V, Ballauf A, Haerynck F, Bordon V, Dhooge C, Garcia-Lloret ML, Bredius RG, Kalwak K, Haddad E, Seidel MG, Duckers G, Pai SY, Dvorak CC, Ehl S, Locatelli F, Goldman F, Gennery AR, Cowan MJ, Roncarolo MG, Bacchetta R, Primary immune deficiency treatment C, the Inborn Errors Working Party of the European Society for B, Marrow T (2018) Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol 141:1036–49 e5
Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, Segundo GRS, Allenspach EJ, Favre C, Ochs HD, Torgerson TR (2018) Clinical, Immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) Syndrome. Front Immunol 9:2411
Louie RJ, Tan QK, Gilner JB, Rogers RC, Younge N, Wechsler SB, McDonald MT, Gordon B, Saski CA, Jones JR, Chapman SJ, Stevenson RE, Sleasman JW, Friez MJ (2017) Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am J Med Genet A 173:1219–1225
Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, Roncarolo MG, Bacchetta R (2013) CD4(+) T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med 5:215ra174
Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, Le DT, Liu W, Brusko TM, Anderson MS, Rudensky AY, Marson A, Chang HY, Bluestone JA (2019) A mutation in the transcription factor Foxp3 drives T helper 2 effector function in regulatory T cells. Immunity 50:362–77 e6
Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG (2006) Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 116:1713–1722
Passerini L, Olek S, Di Nunzio S, Barzaghi F, Hambleton S, Abinun M, Tommasini A, Vignola S, Cipolli M, Amendola M, Naldini L, Guidi L, Cecconi M, Roncarolo MG, Bacchetta R (2011) Forkhead box protein 3 (FOXP3) mutations lead to increased TH17 cell numbers and regulatory T-cell instability. J Allergy Clin Immunol 128:1376–9 e1
Lim HW, Hillsamer P, Kim CH (2004) Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J Clin Invest 114:1640–1649
Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, Fan HM, Liu ZM, Neelapu SS, Dong C (2011) Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17:983–988
Ramiscal RR, Vinuesa CG (2013) T-cell subsets in the germinal center. Immunol Rev 252:146–155
Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY (2007) Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445:771–775
Kuczma M, Podolsky R, Garge N, Daniely D, Pacholczyk R, Ignatowicz L, Kraj P (2009) Foxp3-deficient regulatory T cells do not revert into conventional effector CD4+ T cells but constitute a unique cell subset. J Immunol 183:3731–3741
Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA (2007) Regulatory T cell development in the absence of functional Foxp3. Nat Immunol 8:359–368
Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, Albarello L, Mariani A, Testoni PA, Bazzigaluppi E, Bosi E, Lampasona V, Neth O, Zama D, Hoenig M, Schulz A, Seidel MG, Rabbone I, Olek S, Roncarolo MG, Cicalese MP, Aiuti A, Bacchetta R (2020) Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol 145:1262–71 e13
Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, Garcia-Lloret MI, Chen K, Ozen A, Carmeliet P, Li MO, Pellegrini M, Chatila TA (2019) Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol 20:1208–1219
Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, Jones R, Ring SM, McArdle W, Pembrey ME, Strachan DP, Dunger DB, Twells RC, Clayton DG, Todd JA (2005) Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet 76:773–779
Bakhtiar S, Fekadu J, Seidel MG, Gambineri E (2019) Allogeneic hematopoietic stem cell transplantation for congenital immune dysregulatory disorders. Front Pediatr 7:461v
Walker LS (2013) Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun 45:49–57
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schaffer AA, Gruning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LSK, Sansom DM, Grimbacher B (2014) Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20:1410–1416
Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, Massaad MJ, Garcia-Lloret M, Hanna-Wakim R, Dbaibo G, Alangari AA, Alsultan A, Al-Zahrani D, Geha RS, Chatila TA (2015) Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol 135:217–227
Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, Hoshino A, Kobayashi M, Imai K, Takagi M, Dybedal I, Haddock JA, Sansom DM, Lucena JM, Seidl M, Schmitt-Graeff A, Reiser V, Emmerich F, Frede N, Bulashevska A, Salzer U, Schubert D, Hayakawa S, Okada S, Kanariou M, Kucuk ZY, Chapdelaine H, Petruzelkova L, Sumnik Z, Sediva A, Slatter M, Arkwright PD, Cant A, Lorenz HM, Giese T, Lougaris V, Plebani A, Price C, Sullivan KE, Moutschen M, Litzman J, Freiberger T, van de Veerdonk FL, Recher M, Albert MH, Hauck F, Seneviratne S, Pachlopnik Schmid J, Kolios A, Unglik G, Klemann C, Speckmann C, Ehl S, Leichtner A, Blumberg R, Franke A, Snapper S, Zeissig S, Cunningham-Rundles C, Giulino-Roth L, Elemento O, Duckers G, Niehues T, Fronkova E, Kanderova V, Platt CD, Chou J, Chatila TA, Geha R, McDermott E, Bunn S, Kurzai M, Schulz A, Alsina L, Casals F, Deya-Martinez A, Hambleton S, Kanegane H, Tasken K, Neth O, Grimbacher B (2018) Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 142:1932–1946
Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, Hagin D, Kennedy A, Barge D, Flood T, Abinun M, Wynn RF, Gennery AR, Cant AJ, Sansom D, Hambleton S, Torgerson TR (2016) Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol 138:615–9 e1
Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, Karakoc-Aydiner E, Riviere JG, Garcia-Prat M, Moes N, Haerynck F, Gonzales-Granado LI, Santos Perez JL, Mukhina A, Shcherbina A, Aghamohammadi A, Hammarstrom L, Dogu F, Haskologlu S, Ikinciogullari AI, Kostel Bal S, Baris S, Kilic SS, Karaca NE, Kutukculer N, Girschick H, Kolios A, Keles S, Uygun V, Stepensky P, Worth A, van Montfrans JM, Peters AMJ, Meyts I, Adeli M, Marzollo A, Padem N, Khojah AM, Chavoshzadeh Z, Avbelj Stefanija M, Bakhtiar S, Florkin B, Meeths M, Gamez L, Grimbacher B, Seppanen MRJ, Lankester A, Gennery AR, Seidel MG, Inborn Errors C, Registry Working Parties of the European Society for B, Marrow T, the European Society for I (2020) Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol 145:1452–1463
Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, Passerini L, Narula M, Sheikali A, Lee CM, Bao G, Bauer CS, Miller HK, Garcia-Lloret M, Butte MJ, Bertaina A, Shah A, Pavel-Dinu M, Hendel A, Porteus M, Roncarolo MG, Bacchetta R (2020) CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv 6:eaaz0571
Masiuk KE, Laborada J, Roncarolo MG, Hollis RP, Kohn DB (2019) Lentiviral gene therapy in HSCs restores lineage-specific Foxp3 expression and suppresses autoimmunity in a mouse model of IPEX syndrome. Cell Stem Cell 24:309–17 e7
Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, Tobin V, Nguyen DN, Lee MR, Putnam AL, Ferris AL, Chen JW, Schickel JN, Pellerin L, Carmody D, Alkorta-Aranburu G, Del Gaudio D, Matsumoto H, Morell M, Mao Y, Cho M, Quadros RM, Gurumurthy CB, Smith B, Haugwitz M, Hughes SH, Weissman JS, Schumann K, Esensten JH, May AP, Ashworth A, Kupfer GM, Greeley SAW, Bacchetta R, Meffre E, Roncarolo MG, Romberg N, Herold KC, Ribas A, Leonetti MD, Marson A (2018) Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559:405–409
Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D (2005) The cellular mechanism of Aire control of T cell tolerance. Immunity 23:227–239
Markert ML, Devlin BH, McCarthy EA (2010) Thymus transplantation. Clin Immunol 135:236–246
Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, Thamsen M, Santos-Cortez RL, Lee K, Gambin T, Forbes LR, Law CS, Stray-Pedersen A, Cheng MH, Mace EM, Anderson MS, Liu D, Tang LF, Nicholas SK, Nahmod K, Makedonas G, Canter DL, Kwok PY, Hicks J, Jones KD, Penney S, Jhangiani SN, Rosenblum MD, Dell SD, Waterfield MR, Papa FR, Muzny DM, Zaitlen N, Leal SM, Gonzaga-Jauregui C, Baylor-Hopkins Center for Mendelian G, Boerwinkle E, Eissa NT, Gibbs RA, Lupski JR, Orange JS, Shum AK. (2015) COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 47:654–660
Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, Eldin KW, Bertolet G, McKinley S, de Guzman M, Forbes L, Chinn I, Orange JS (2016) Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol 36:377–387
Deng Z, Law CS, Ho FO, Wang KM, Jones KD, Shin JS, Shum AK (2020) A defect in thymic tolerance causes T cell-mediated autoimmunity in a murine model of COPA syndrome. J Immunol 204:2360–2373
Lepelley A, Martin-Niclos MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, Bondet V, Duffy D, Hertzog J, Rehwinkel J, Amselem S, Boulisfane-El Khalifi S, Brennan M, Carter E, Chatenoud L, Chhun S, Coulomb l’Hermine A, Depp M, Legendre M, Mackenzie KJ, Marey J, McDougall C, McKenzie KJ, Molina TJ, Neven B, Seabra L, Thumerelle C, Wislez M, Nathan N, Manel N, Crow YJ, Fremond ML (2020) Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J Exp Med 217:e20200600
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36:955–957
Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, Montgomery T, Goodship JA, Burt AD, Flood TJ, Abinun M, Cant AJ, Johnson D (2008) Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol 153:75–80
Guris DL, Duester G, Papaioannou VE, Imamoto A (2006) Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell 10:81–92
Zemble R, Luning Prak E, McDonald K, McDonald-McGinn D, Zackai E, Sullivan K (2010) Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol 136:409–418
Deshpande DR, Demirdag YY, Marsh RA, Sullivan KE, Orange JS, Consortium U. 2020. Relationship between severity of T cell lymphopenia and immune dysregulation in patients with DiGeorge syndrome (22q11.2 Deletions and/or Related TBX1 Mutations): a USIDNET Study. J Clin Immunol
Staple L, Andrews T, McDonald-McGinn D, Zackai E, Sullivan KE (2005) Allergies in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) and patients with chronic granulomatous disease. Pediatr Allergy Immunol 16:226–230
Di Cesare S, Puliafito P, Ariganello P, Marcovecchio GE, Mandolesi M, Capolino R, Digilio MC, Aiuti A, Rossi P, Cancrini C (2015) Autoimmunity and regulatory T cells in 22q11.2 deletion syndrome patients. Pediatr Allergy Immunol 26:591–594
Marcovecchio GE, Bortolomai I, Ferrua F, Fontana E, Imberti L, Conforti E, Amodio D, Bergante S, Macchiarulo G, D’Oria V, Conti F, Di Cesare S, Fousteri G, Carotti A, Giamberti A, Poliani PL, Notarangelo LD, Cancrini C, Villa A, Bosticardo M (2019) Thymic epithelium abnormalities in DiGeorge and Down syndrome patients contribute to dysregulation in T cell development. Front Immunol 10:447
Chinn IK, Milner JD, Scheinberg P, Douek DC, Markert ML (2013) Thymus transplantation restores the repertoires of forkhead box protein 3 (FoxP3)+ and FoxP3- T cells in complete DiGeorge anomaly. Clin Exp Immunol 173:140–149
Ucar O, Tykocinski LO, Dooley J, Liston A, Kyewski B (2013) An evolutionarily conserved mutual interdependence between Aire and microRNAs in promiscuous gene expression. Eur J Immunol 43:1769–1778
Khan IS, Taniguchi RT, Fasano KJ, Anderson MS, Jeker LT (2014) Canonical microRNAs in thymic epithelial cells promote central tolerance. Eur J Immunol 44:1313–1319
Bernard C, Frih H, Pasquet F, Kerever S, Jamilloux Y, Tronc F, Guibert B, Isaac S, Devouassoux M, Chalabreysse L, Broussolle C, Petiot P, Girard N, Seve P (2016) Thymoma associated with autoimmune diseases: 85 cases and literature review. Autoimmun Rev 15:82–92
Cheng MH, Fan U, Grewal N, Barnes M, Mehta A, Taylor S, Husebye ES, Murphy EJ, Anderson MS (2010) Acquired autoimmune polyglandular syndrome, thymoma, and an AIRE defect. N Engl J Med 362:764–766
Ferre EMN, Break TJ, Burbelo PD, Allgauer M, Kleiner DE, ** D, Xu Z, Folio LR, Mollura DJ, Swamydas M, Gu W, Hunsberger S, Lee CR, Bondici A, Hoffman KW, Lim JK, Dobbs K, Niemela JE, Fleisher TA, Hsu AP, Snow LN, Darnell DN, Ojaimi S, Cooper MA, Bozzola M, Kleiner GI, Martinez JC, Deterding RR, Kuhns DB, Heller T, Winer KK, Rajan A, Holland SM, Notarangelo LD, Fennelly KP, Olivier KN, Lionakis MS. 2019. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med 11
Wolff AS, Karner J, Owe JF, Oftedal BE, Gilhus NE, Erichsen MM, Kampe O, Meager A, Peterson P, Kisand K, Willcox N, Husebye ES (2014) Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J Immunol 193:3880–3890
Hapnes L, Willcox N, Oftedal BE, Owe JF, Gilhus NE, Meager A, Husebye ES, Wolff AS (2012) Radioligand-binding assay reveals distinct autoantibody preferences for type I interferons in APS I and myasthenia gravis subgroups. J Clin Immunol 32:230–237
Ferrara JL, Levine JE, Reddy P, Holler E (2009) Graft-versus-host disease. Lancet 373:1550–1561
Ukena SN, Grosse J, Mischak-Weissinger E, Buchholz S, Stadler M, Ganser A, Franzke A (2011) Acute but not chronic graft-versus-host disease is associated with a reduction of circulating CD4(+)CD25 (high)CD127 (low/-) regulatory T cells. Ann Hematol 90:213–218
Seemayer TA, Lapp WS, Bolande RP (1977) Thymic involution in murine graft-versus-host reaction. Epithelial injury mimicking human thymic dysplasia. Am J Pathol 88:119–134
Seddik M, Seemayer TA, Lapp WS (1980) T cell functional defect associated with thymid epithelial cell injury induced by a graft-versus-host reaction. Transplantation 29:61–66
Rossi S, Blazar BR, Farrell CL, Danilenko DM, Lacey DL, Weinberg KI, Krenger W, Hollander GA (2002) Keratinocyte growth factor preserves normal thymopoiesis and thymic microenvironment during experimental graft-versus-host disease. Blood 100:682–691
Dertschnig S, Hauri-Hohl MM, Vollmer M, Hollander GA, Krenger W (2015) Impaired thymic expression of tissue-restricted antigens licenses the de novo generation of autoreactive CD4+ T cells in acute GVHD. Blood 125:2720–2723
Funding
The authors received support from the Center for Genetic Immune Diseases (CGID) and the Center for Definitive and Curative Medicine at Stanford University, and a gift from the Lucile Packard Foundation for Children’s Health. Dr. Bacchetta is an Anne T. and Robert M. Bass Faculty Scholar of the Department of Pediatrics, Stanford University School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on: The thymus and autoimmunity - Guest Editor: Georg Holländer
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bacchetta, R., Weinberg, K. Thymic origins of autoimmunity—lessons from inborn errors of immunity. Semin Immunopathol 43, 65–83 (2021). https://doi.org/10.1007/s00281-020-00835-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-020-00835-8