
Abstract
Aims/hypothesis
Associations have been described between higher birthweight and increased risk of type 1 diabetes, and of insulin (INS) and human leucocyte antigen (HLA) genotypes that protect against diabetes with larger size at birth. We studied simultaneously the effects of size at birth, INS and HLA genotypes on the risk of type 1 diabetes to test whether the relation between size at birth and risk of type 1 diabetes would be strengthened after adjustment for INS and HLA genotypes.
Subjects and methods
We designed a population-based case–control study in Norway with 471 cases of childhood-onset type 1 diabetes and 1,369 control subjects who were genotyped for the INS -23HphI polymorphism (surrogate for INS variable number of tandem repeats) and HLA-DQ alleles associated with type 1 diabetes. Data on birthweight and other perinatal factors were obtained from the Medical Birth Registry of Norway by record linkage.
Results
The data fitted a multiplicative model for the protective INS class III allele both within the INS locus and for the model with INS- and HLA-DQ-conferred risk of type 1 diabetes. We found no overall significant association between weight or head circumference at birth and the risk of type 1 diabetes, and adjustment for INS and HLA genotype did not influence this result. There was also no evidence for association of INS or HLA with size at birth among control subjects.
Conclusions/interpretation
In contrast to suggestions from previous indirect studies, direct adjustment for INS and HLA genotypes did not lead to a stronger relation between birthweight and the risk of type 1 diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes is a result of immune-mediated destruction of the pancreatic beta cells, but the factors initiating the destructive process are largely unknown [1]. There is evidence for the involvement of both genetic and non-genetic factors in this development. The two quantitatively most important genetic susceptibility loci influencing the risk of type 1 diabetes are the HLA class II region and the insulin gene (INS) [2]. Several HLA-DQ and -DR alleles and genotypes are associated with risk and protection to different degrees [3], and the INS variable number of tandem repeats (VNTR) class I or its surrogate marker, the -23HphI A allele, has consistently been found to confer increased risk of type 1 diabetes [4, 5]. Accumulating evidence suggests that environmental factors may play a role early in life [6] and that there is an effect of size at birth and other perinatal factors on the risk of type 1 diabetes, generally of relatively small magnitude [7–11]. Several large-scale studies have found a high birthweight to be associated with a modest but statistically significant increase in the risk of childhood-onset type 1 diabetes [8, 9, 11], although other, predominately smaller studies have not found a significant association [e.g. 7, 12]. The mechanism by which INS VNTR class III (or the linked -23HphI T) allele reduces the risk of type 1 diabetes has been hypothesised to be via increased thymic expression of insulin during fetal and/or postnatal life [4]. INS polymorphisms may also affect fetal growth via a variety of pathways, including differential expression of IGF2, although fetal pancreatic insulin expression is only moderately affected by allelic variation at the INS VNTR [4]. Some studies of children without diabetes have indicated that type 1 diabetes-protective alleles at the INS and HLA loci are associated with larger size at birth [13–16], suggesting that any positive association between size at birth and the risk of type 1 diabetes may be attenuated by allelic variation at these loci. If so, the association between size at birth and the risk of type 1 diabetes should be strengthened after statistical adjustment for variation at these loci.
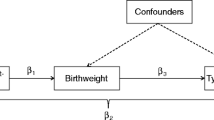
We therefore aimed to assess directly whether the odds ratio for the relation between size at birth and type 1 diabetes would be strengthened after adjustment for the INS -23HphI polymorphism and HLA-DQ genotypes, regardless of whether an association existed before adjustment. Secondary objectives were to assess the joint effects of HLA-DQ and INS polymorphism on the risk of type 1 diabetes, associations of INS and HLA with size at birth in the general population, and the role of birth order in these relations.
Subjects and methods
Study design
We designed a population-based case–control study in Norway, using cases of type 1 diabetes diagnosed before age 15 years between 1997 and 2000 and control children in the same age group randomly selected from the official population registry. All children were born between 1985–1999 and aged 1–15 years at the time of data collection [17]. Fifty-six per cent of controls and 73% of cases participated, with similar participation rates among regions, sexes and age groups [18]. DNA was collected using mailed mouth swabs and extracted as previously described [19]. Information on perinatal factors was obtained from the Medical Birth Registry of Norway [20] by record linkage and additional data were collected using questionnaires mailed to the families [17]. Written informed consent was obtained from all participants, and the regional ethics committee and the Norwegian Data Inspectorate approved the study. After exclusion of children not born in Norway, twins, and children whose mother had diabetes during pregnancy, a total of 471 cases and 1,369 controls had complete data on INS, HLA and size at birth.
Genoty**
For both HLA and INS genoty**, cases and controls were genotyped at the same time, and laboratory personnel were blinded to the status (case or control) of the DNA samples. HLA genoty** was performed using sequence-specific PCR identifying HLA-DQA1*02, DQA1*03, DQA1*05, HLA-DQB1*0201, DQB1*0201/0302, DQB1*0302/3, DQB1*0303, DQB1*0602, DQB1*0603/7 and DQB1*0301/4, as previously described [21, 22]. DQA1-DQB1 haplotypes were determined based on known linkage equilibria. Samples identified as DQA1*05-DQB1*0201/X (DQ2/X) were typed for additional alleles to determine whether they were homozygous for DQ2 using PCR and allele-specific lanthanide (III) chelate-labelled probes in streptavidin-coated microtitration wells according to the manufacturer’s protocol (Perkin-Elmer Life Sciences, Turku, Finland). Individuals were categorised in four groups based on HLA type: (1) protective: at least one HLA-DQB1*0602 allele; (2) high risk: DQA1*03-DQB1*0302/DQA1*05-DQB1*0201 (DQ8/DQ2); (3) moderate risk: DQ2/DQ2, DQ8/DQ8 or DQ8/X, where X represents alleles/haplotypes not defined above; and (4) all other genotypes (X/X, DQ2/X), which were regarded as a neutral reference category for the purpose of this study, although this latter category includes genotypes with some variation in risk.
The INS -23HphI polymorphism (surrogate for INS VNTR) was genotyped in two independent laboratories. One laboratory used a TaqMan assay (Applied Biosystems, Foster City, CA, USA), run on an ABI 7900 HT Fast Real-time PCR system (Applied Biosystems). The other laboratory used RFLP analysis as previously described [23], but using nested PCR. Discrepant results were re-run in both laboratories and resolved or excluded. Additional details on genoty** are available from the authors. We refer to the -23HphI A (risk) allele as (VNTR) class I and the T (protective) allele as class III [4].
Data analysis
We used logistic regression analysis to assess associations of size at birth, INS and HLA genotypes with type 1 diabetes, including likelihood ratio tests of multiplicative interactions, and we used ANOVA and linear regression to assess associations of INS and HLA genotypes with size at birth among control children. A two-sided p value less than 0.05 or a 95% CI excluding the null value (1.00 for the odds ratio) was taken as indicating statistical significance.
Power
Based on previous results [9], we assumed for the prestudy power analyses a logit-linear model for the risk of type 1 diabetes according to birthweight in four groups, and that the crude odds ratio for birth weight of 4,000 g or greater vs <2,500 g is 1.6. We hypothesised that the odds ratio would increase to 1.8 after adjustment for INS and HLA genotype. This scenario would provide 81% power to detect a significant association between birthweight and type 1 diabetes with 500 cases and 1,500 controls. Under a recessive model (INS class III or I/III vs I/I), the use of 1,500 control subjects provides 90% power to detect significant differences in birthweight and head circumference if the true difference is 89 g and 0.27 cm, respectively.
Results
Perinatal factors were not significantly associated with type 1 diabetes, but there was a slightly lower risk for children whose mothers were better educated, as previously reported [17] (Electronic supplementary material [ESM] Table 1). The well-established effect of HLA on the risk of type 1 diabetes was confirmed; the distribution among the 471 cases was 8, 83, 208 and 172 with protective, neutral, moderate-risk and high-risk genotypes, respectively, and 412, 619, 281 and 57, respectively, among the 1,369 controls (ESM Table 2). The effect of HLA was essentially similar after stratification by birth order, with no significant interaction (not shown).
The distribution of INS genotypes class III/III, I/III and I/I was 15, 120 and 336 among cases and 111, 527 and 731 among controls. The INS data fitted a multiplicative odds model (gene dose model) very well, with a nearly two-fold decrease in the risk of type 1 diabetes per protective class III allele (ESM Table 2). The odds ratio for the class I/I genotype vs I/III or III/III was 1.75 for first-born children and 2.55 for second- and later-born children, but the interaction between birth order and INS was not significant (p=0.11).
The odds ratio for INS class I/I vs I/III or III/III was very similar in all subgroups of HLA-associated risk, with no significant deviation from a multiplicative model for INS- and HLA-conferred risk (ESM Table 3).
Association between size at birth and type 1 diabetes after adjustment for INS and HLA
There was a tendency for those with the lowest birthweight to have a higher risk of type 1 diabetes, but there was no overall significant association between birthweight and the risk of type 1 diabetes, and this was essentially not influenced by adjustment for INS, HLA or other potential confounders (Table 1). No significant interaction between birthweight and INS (p=0.83), HLA (p=0.62) or birth order (p=0.28) was observed. Very similar patterns were found for head circumference at birth (Table 1).
Association of INS and HLA with size at birth among population-based control children
There were significant associations of sex, gestational age, birth order and maternal smoking during pregnancy with size at birth (ESM Table 4). There was no significant association of INS genotype with size at birth (Table 2). No association was seen in either category of birth order (not shown).
Children carrying the type 1 diabetes-protective allele HLA-DQB1*0602 tended to be larger at birth, but the overall ANOVA test was not significant (Table 2), and this suggestive association was attenuated after adjustment for covariates (not shown). Inspired by a recent report [16], we also investigated HLA-DQB1*0603. The 170 control children carrying at least one HLA-DQB1*0603 allele had a mean birthweight of 3,594 g compared with 3,621 g for non-carriers (p=0.52).
Discussion
We present for the first time a simultaneous analysis of the effect of size at birth and genetic factors on the risk of type 1 diabetes in a well-powered study. We were thus able to test directly whether the association between size at birth and type 1 diabetes is enhanced after adjustment for INS and HLA genotypes. Although previous indirect studies have provided circumstantial evidence for such enhancement [13–16], our direct test in the present report showed that this was not the case. The controls in the present study included only individuals who were not part of our pilot study [15], and thus provide an independent study of controls. Of the 471 cases, 175 (37%) were also part of our previous cohort study based on linkage of registries without information on molecular genetic markers [9], whereas the rest of the cases in the present study had not been analysed previously.
Strengths of the present study include: (1) the inclusion of children drawn from registries with nearly complete population coverage; (2) linkage of registries via unique personal identification numbers; (3) the inclusion of a large number of type 1 diabetes cases; and (4) adjustment for a number of relevant covariates.
Among the study’s limitations were: (1) we did not know the parental origin of the INS and HLA alleles; (2) we did not have postnatal growth data; and (3) participation was incomplete — a problem inherent in most clinical research. Although the response in our study was in line with that in many other studies in which biological specimens were collected [24], there is room for potential bias. The distribution of birthweight among the controls in the present study was shifted towards higher birthweights compared to the distribution of birthweights reported to The Medical Birth Registry of Norway [20] between 1985 and 1998 in which the mother did not have diabetes registered before or during pregnancy (e.g. 1.6% of participating controls had a birthweight <2,500 g compared with 4.5% for all births in The Medical Birth Registry). Our controls also had mothers who were slightly older when giving birth compared with the background population. This may have been due to selection of participating controls whose mothers were more highly educated than average. Regarding maternal education, Vangen et al. [25] linked data from Statistics Norway on maternal education to The Medical Birth Registry of Norway for all births during 1986–1998, and found that 30.5% of women had more than 12 years of education, 59.8% had 10–12 years of education, 8.4% had less than 10 years, and 1.3% had missing information. Corresponding results from our participating control children [18] were 37.1% (>12 years), 54.3% (10–12 years), 7.3% (<10 years) and 1.4% (missing information). Despite slightly different calendar years and inclusion criteria, the above data indicate selection of children with higher birthweight and older and more highly educated mothers among our controls. Whether similar selection among our cases with type 1 diabetes occurred is unknown, but we can speculate that any such selection may have been weaker among case children, in whom there was a higher proportion of responses. If so, this may partly explain the lack of association of maternal age and birthweight with the risk of type 1 diabetes in the present study in contrast to our earlier reports [9, 10], in addition to the smaller sample size in the present study. On the other hand, there is little reason to suspect that the effect of molecular genetic markers on size at birth or on the relation between size at birth and type 1 diabetes should be influenced by any such potential bias, because it is hard to imagine that INS or HLA genotype should be strongly associated with behaviour directing participation in the study.
In relation to our secondary objectives, we note that the initial report of an association between the INS polymorphism and size at birth among children from the general population [13] has not been supported by recent reports [26–28]. A new study from the UK has indicated that the association between INS and size at birth primarily concerns head circumference, and was stronger for second and later children than for first-born children, presumably because first-born children experience more environmental restrictions on fetal growth than later-born children [14]. In our study, we found no indication of association between INS and size at birth in any group of birth order. In a previous study [15], we found a tendency for the HLA-DQB1*0602 allele, which is strongly protective against type 1 diabetes, to be associated with higher birthweight. We found a similar indication in the present data set, but this was not a significant or robust finding. A very large study from Sweden reported that HLA-DQB1*0603 (protective against type 1 diabetes) and the high-risk DQ2/DQ8 genotype were associated with higher birthweight for gestational age [16], but we were unable to confirm this in our study. Consistent with other recent results [5], the protective class III alleles seemed to display a multiplicative allele dose effect, in contrast to the previous notion of a dominantly protective effect against type 1 diabetes [4].
From the present results it seems that, at least in our population, possible effects of INS and HLA on birthweight [13, 15, 16] and of birthweight on the risk of type 1 diabetes [9, 11] are of too small a magnitude to allow clear findings of confounding or interacting effects on the risk of type 1 diabetes unless sample sizes are exceedingly large.
We conclude that, in contrast to indications from previous indirect studies, the relation between size at birth and type 1 diabetes was not influenced by adjustment for INS and HLA.
Abbreviations
- ESM:
-
Electronic supplementary material
- INS :
-
insulin gene
- VNTR:
-
variable number of tandem repeats
References
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Concannon P, Erlich HA, Julier C et al (2005) Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes 54:995–1001
Undlien DE, Lie BA, Thorsby E (2001) HLA complex genes in type 1 diabetes and other autoimmune diseases. Which genes are involved? Trends Genet 17:93–100
Pugliese A, Miceli D (2002) The insulin gene in diabetes. Diabetes Metab Res Rev 18:13–25
Barratt BJ, Payne F, Lowe CE et al (2004) Remap** the insulin gene/Iddm2 locus in type 1 diabetes. Diabetes 53:1884–1889
Leslie DG, Elliott RB (1994) Early environmental events as a cause of IDDM. Diabetes 43:843–850
Blom L, Dahlquist G, Nyström L, Sandström A, Wall S (1989) The Swedish Childhood Diabetes Study—social and perinatal determinants for diabetes in childhood. Diabetologia 32:7–13
Dahlquist G, Patterson C, Stoltesz G (1999) Perinatal risk factors for childhood type 1 diabetes in Europe: the EURODIAB Substudy 2 Study Group. Diabetes Care 22:1698–1702
Stene LC, Magnus P, Lie RT, Søvik O, Joner G, The Norwegian Childhood Diabetes Study Group (2001) Birth weight and childhood onset type 1 diabetes: population based cohort study. BMJ 322:889–892
Stene LC, Magnus P, Lie RT, Søvik O, Joner G, The Norwegian Childhood Diabetes Study Group (2001) Maternal and paternal age at delivery, birth order, and risk of childhood onset type 1 diabetes: population based cohort study. BMJ 323:369–371
Dahlquist GG, Pundziute-Lyckå A, Nyström L (2005) Birthweight and risk of type 1 diabetes in children and young adults: a population-based register study. Diabetologia 48:1114–1117
Kyvik KO, Bache I, Green A, Beck-Nielsen H, Buschard K (2000) No association between birth weight and type 1 diabetes mellitus—a twin-control study. Diabet Med 17:158–162
Dunger DB, Ong KK, Huxtable SJ et al (1998) Association of the INS VNTR with size at birth. ALSPAC study team. Avon longitudinal study of pregnancy and childhood. Nat Genet 19:98–100
Ong KK, Petry CJ, Barratt BJ et al (2004) Maternal–fetal interactions and birth order influence insulin variable number of tandem repeats allele class associations with head size at birth and childhood weight gain. Diabetes 53:128–133
Stene LC, Magnus P, Rønningen KS, Joner G (2001) Diabetes-associated HLA-DQ genes and birth weight. Diabetes 50:2879–2882
Larsson HE, Lynch K, Lernmark B et al (2005) Diabetes-associated HLA genotypes affect birthweight in the general population. Diabetologia 48:1484–1491
Stene LC, Joner G, The Norwegian Childhood Diabetes Study Group (2004) Atopic disorders and risk of childhood-onset type 1 diabetes in individuals. Clin Exp Allergy 34:201–206
Stene LC, Joner G, The Norwegian Childhood Diabetes Study Group (2003) Use of cod liver oil during the first year of life is associated with lower risk of childhood-onset type 1 diabetes: a large, population based, case–control study. Am J Clin Nutr 78:1128–1134
Witsø E, Stene LC, Paltiel L, Joner G, Rønningen KS (2002) DNA extraction and HLA genoty** using mailed mouth brushes from children. Pediatr Diabetes 3:89–94
Irgens LM (2000) The Medical Birth Registry of Norway. Epidemiological research and surveillance throughout 30 years. Acta Obstet Gynecol Scand 79:435–439
Olerup O, Aldener A, Fogdell A (1993) HLA-DQB1 and -DQA1 ty** by PCR amplification with sequence-specific primers (PCR-SSP) in 2 hours. Tissue Antigens 41:119–134
Cinek O, Koloušková S, Šnajderová M et al (2001) HLA class II genetic association of type 1 diabetes mellitus in Czech children. Pediatr Diabetes 2:98–102
Thorsby PM, Berg JP, Birkeland KI (2005) Insulin gene variable number of tandem repeats is associated with increased fat mass during adolescence in non-obese girls. Scand J Clin Lab Invest 65:163–168
Morton LM, Cahill J, Hartge P (2006) Reporting participation in epidemiologic studies: a survey of practice. Am J Epidemiol 163:197–203
Vangen S, Stoltenberg C, Johansen RE, Sundby J, Stray-Pedersen B (2002) Perinatal complications among ethnic Somalis in Norway. Acta Obstet Gynecol Scand 81:317–322
Lindsay RS, Hanson RL, Wiedrich C, Knowler WC, Bennett PH, Baier LJ (2003) The insulin gene variable number tandem repeat class I/III polymorphism is in linkage disequilibrium with birth weight but not type 2 diabetes in the Pima population. Diabetes 52:187–193
Mitchell SM, Hattersley AT, Knight B et al (2004) Lack of support for a role of the insulin gene variable number of tandem repeats minisatellite (INS-VNTR) locus in fetal growth or type 2 diabetes-related intermediate traits in United Kingdom populations. J Clin Endocrinol Metab 89:310–317
Bennett AJ, Sovio U, Ruokonen A et al (2004) Variation at the insulin gene VNTR (variable number tandem repeat) polymorphism and early growth: studies in a large Finnish birth cohort. Diabetes 53:2126–2131
Acknowledgements
L. C. Stene and G. Joner were supported by a grant from The Research Council of Norway (148359/330). Funding for L. C. Stene and G. Joner was also provided by the Norwegian Diabetes Association, Aker Diabetes Research Foundation, Novo Nordisk Fonden, Peter Möller AS, TINE Norwegian Diaries and Novo Nordisk Scandinavia. J. P. Berg and D. E. Undlien were supported by Eastern Regional Health Authorities. The latter was also supported by The National Programme for Research in Functional Genomics in Norway (FUGE) of the Research Council of Norway. We thank the staff at the Medical Birth Registry of Norway for their assistance with record linkage and the staff at the Biobank, Norwegian Institute of Public Health, for assistance with DNA extraction and genoty**. Specifically we thank H. E. Akselsen, M. Bjørnvold and K. Gervin at the Institute for Medical Genetics, Ullevål University Hospital, and V. Enge at Hormone Laboratory, Aker University Hospital, for help with genoty**.
Duality of interest
The authors declare that they have no duality of interest.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Members of Norwegian Childhood Diabetes Study Group, see Appendix
Appendix
Members of the Norwegian Childhood Diabetes Study Group
Henning Aabech, Fredrikstad; Helge Vogt, Nordbyhagen; Knut Dahl-Jørgensen, Oslo; Kolbeinn Gudmundsson, Oslo; Jon Grøtta, Elverum; Halvor Bævre, Gjøvik; Ola Talleraas, Lillehammer; Kjell Stensvold, Drammen; Bjørn Halvorsen, Tønsberg; Kristin Hodnekvam, Skien; Ole Kr. Danielsen, Arendal; Jorunn Ulriksen, Kristiansand; John Bland, Stavanger; Dag Roness, Haugesund; Oddmund Søvik and Pål R. Njølstad, Bergen; Per Helge Kvistad, Førde; Steinar Spangen, Ålesund; Per Eirik Hæreid, Trondheim; Sigurd Børsting, Levanger; Dag Veimo, Bodø; Harald Dramsdahl, Harstad; Bård Forsdahl, Tromsø; Kersti Thodenius, Hammerfest.
Electronic supplementary material
Below is the link of the electronic supplementary material.
Supplementary Table 1
(PDF 40 kb)
Supplementary Table 2
(PDF 50 kb)
Supplementary Table 3
(PDF 46 kb)
Supplementary Table 4
(PDF 41 kb)
Rights and permissions
About this article
Cite this article
Stene, L.C., Thorsby, P.M., Berg, J.P. et al. The relation between size at birth and risk of type 1 diabetes is not influenced by adjustment for the insulin gene (-23HphI) polymorphism or HLA-DQ genotype. Diabetologia 49, 2068–2073 (2006). https://doi.org/10.1007/s00125-006-0292-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0292-6