Search
Search Results
-
Insights into the enumeration of mixtures of probiotic bacteria by flow cytometry
The use of flow cytometry to enumerate microorganisms is gaining traction over the traditional plate count technique on the basis of superior...

-
EFMlrs: a Python package for elementary flux mode enumeration via lexicographic reverse search
BackgroundElementary flux mode (EFM) analysis is a well-established, yet computationally challenging approach to characterize metabolic networks....

-
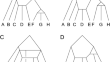
All galls are divided into three or more parts: recursive enumeration of labeled histories for galled trees
ObjectiveIn mathematical phylogenetics, a labeled rooted binary tree topology can possess any of a number of labeled histories, each of which...
-
Embedding gene trees into phylogenetic networks by conflict resolution algorithms
BackgroundPhylogenetic networks are mathematical models of evolutionary processes involving reticulate events such as hybridization, recombination,...

-
Tree diet: reducing the treewidth to unlock FPT algorithms in RNA bioinformatics
Hard graph problems are ubiquitous in Bioinformatics, inspiring the design of specialized Fixed-Parameter Tractable algorithms, many of which rely on...

-
A Low-Cost Proximate Sensing Method for Early Detection of Nematodes in Walnut Using Machine Learning Algorithms
This chapter presents an innovative low-cost proximate sensing method designed for the early detection of nematodes in walnut trees, leveraging...
-
Heuristic algorithms for best match graph editing
BackgroundBest match graphs (BMGs) are a class of colored digraphs that naturally appear in mathematical phylogenetics as a representation of the...

-
CombFold: predicting structures of large protein assemblies using a combinatorial assembly algorithm and AlphaFold2
Deep learning models, such as AlphaFold2 and RosettaFold, enable high-accuracy protein structure prediction. However, large protein complexes are...

-
Learning self-supervised molecular representations for drug–drug interaction prediction
Drug–drug interactions (DDI) are a critical concern in healthcare due to their potential to cause adverse effects and compromise patient safety....

-
Algorithms for the quantitative Lock/Key model of cytoplasmic incompatibility
Cytoplasmic incompatibility (CI) relates to the manipulation by the parasite Wolbachia of its host reproduction. Despite its widespread occurrence,...

-
Heuristic shortest hyperpaths in cell signaling hypergraphs
BackgroundCell signaling pathways, which are a series of reactions that start at receptors and end at transcription factors, are basic to systems...

-
Automated design of dynamic programming schemes for RNA folding with pseudoknots
Although RNA secondary structure prediction is a textbook application of dynamic programming (DP) and routine task in RNA structure analysis, it...

-
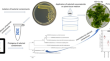
Elimination of Curtobacterium sp. strain A7_M15, a contaminant in Prunus rootstock tissue culture production, using reduced graphene oxide–silver–copper and silver–selenium nanocomposites
BackgroundBacterial contamination poses a high risk to the successful establishment and maintenance of plant tissue cultures. The aim of this study...
-
Infrared: a declarative tree decomposition-powered framework for bioinformatics
MotivationMany bioinformatics problems can be approached as optimization or controlled sampling tasks, and solved exactly and efficiently using...

-
Integrating transformers and many-objective optimization for drug design
BackgroundDrug design is a challenging and important task that requires the generation of novel and effective molecules that can bind to specific...

-
An SMT-Based Framework for Reasoning About Discrete Biological Models
We present a framework called the Reasoning Engine, which implements Satisfiability Modulo Theories (SMT) based methods within a unified...
-
Efficiently sparse listing of classes of optimal cophylogeny reconciliations
BackgroundCophylogeny reconciliation is a powerful method for analyzing host-parasite (or host-symbiont) co-evolution. It models co-evolution as an...

-
Technologies Promoting Genome-Based Taxonomy
Whole-genome sequencing (WGS) has proven to be a reliable method. Additionally, it is a commonly used approach in research and surveillance...
-
RASMA: a reverse search algorithm for mining maximal frequent subgraphs
BackgroundGiven a collection of coexpression networks over a set of genes, identifying subnetworks that appear frequently is an important research...

-
Speeding up the core algorithm for the dual calculation of minimal cut sets in large metabolic networks
BackgroundThe concept of minimal cut sets (MCS) has become an important mathematical framework for analyzing and (re)designing metabolic networks....
