Search
Search Results
-
Relativistic Quantum Chemical and Molecular Dynamics Techniques for Medicinal Chemistry of Bioinorganic Compounds
In this chapter we review relativistic quantum chemical and molecular dynamics techniques focused on drug discovery and predictive toxicology...
-
Structure-Based Design of Epigenetic Inhibitors
Computer-aided and structure-based design methods play an important role in the development of inhibitors for epigenetic drug targets. Multiple hits...
-
Relationship between the binding free energy and PCBs’ migration, persistence, toxicity and bioaccumulation using a combination of the molecular docking method and 3D-QSAR
The molecular docking method was used to calculate the binding free energies between biphenyl dioxygenase and 209 polychlorinated biphenyl (PCB)...

-
Relation between structures of naphthalenylchalcone derivatives and their cytotoxic effects on HCT116 human colon cancer cells
To find potent chemotherapeutic agents, cytotoxic effects of 42 synthetic chalcone derivatives bearing naphthyl groups on HCT116 human colon cancer...

-
Prediction of different antibacterial activity in a new set of formyl hydroxyamino derivatives with potent action on peptide deformylase using structural information
Due to the essential role of peptide deformylase (PDF) at the bacterial growth cycle, it is a noteworthy target for develo** a novel antibacterial...

-
Cheminformatics Explorations of Natural Products
The chemistry of natural products is fascinating and has continuously attracted the attention of the scientific community for many reasons including,...
-
Quantitative surface field analysis: learning causal models to predict ligand binding affinity and pose
We introduce the QuanSA method for inducing physically meaningful field-based models of ligand binding pockets based on structure-activity data...
-
Computer-aided prediction of biological activity spectra for organic compounds: the possibilities and limitations
We describe the current version of the PASS program for prediction of biological activity spectra of organic compounds based on analysis of...
-
Systematic Review on Cytotoxic and Anticancer Potential of N-Substituted Isatins as Novel Class of Compounds Useful in Multidrug-Resistant Cancer Therapy: In Silico and In Vitro Analysis
AbstractAs the emergence of resistance to clinical cancer treatments poses a significant problem in cancer management, there is a constant need to...

-
Semi-correlations as a tool to build up categorical (active/inactive) model of GABAA receptor modulator activity
The interpretation of mode of action for GABA A receptor modulator activity is an important task of medicinal chemistry. The computational elucidation...

-
Pharmacophore interactions analysis and prediction of inhibitory activity of 1,7-diazacarbazoles as checkpoint kinase 1 inhibitors: application of molecular docking, 3D-QSAR and RBF neural network
In the present study, we mainly focused on new synthesized 1,7-diazacarbazole derivatives (44 active molecules) as Chk1 inhibitors to build 3D-QSAR...

-
Molecular insight into the interaction mechanisms of an annulated pyrazole (DB08446) with HIV-1 RT: a QM and QM/QM′ study
AbstractQuantum mechanical and hybrid QM/QM′ ONIOM methodology have been applied to investigate the structure and molecular interactions between...

-
Exploring non-linear distance metrics in the structure–activity space: QSAR models for human estrogen receptor
BackgroundQuantitative structure-activity relationship (QSAR) models are important tools used in discovering new drug candidates and identifying...

-
Combined pharmacophore-guided 3D-QSAR, molecular docking, and virtual screening on bis-benzimidazoles and ter-benzimidazoles as DNA–topoisomerase I poisons
Certain DNA minor groove binders, especially bis-benzimdazole containing compounds, such as Hoechst 33258 and its derivatives, act as potent...

-
Towards reproducible computational drug discovery
The reproducibility of experiments has been a long standing impediment for further scientific progress. Computational methods have been instrumental...

-
Recent Developments in 3D QSAR and Molecular Docking Studies of Organic and Nanostructures
The development of quantitative structure–activity relationship (QSAR) methods is going very fast for the last decades. OSAR approach already plays...
-
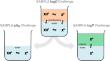
Assessing the accuracy of octanol–water partition coefficient predictions in the SAMPL6 Part II log P Challenge
The SAMPL Challenges aim to focus the biomolecular and physical modeling community on issues that limit the accuracy of predictive modeling of...
-
Molecular docking, MM/GBSA and 3D-QSAR studies on EGFR inhibitors
Epidermal growth factor receptor (EGFR) is the first growth factor receptor proposed as a target for cancer therapy. Molecular modeling protocols...

-
On Applications of QSARs in Food and Agricultural Sciences: History and Critical Review of Recent Developments
During the past decade, a large number of reports described the roles of in silicoIn silico approaches in the development of new molecules...
-
Hybrid caffeic acid derivatives as monoamine oxidases inhibitors: synthesis, radical scavenging activity, molecular docking studies and in silico ADMET analysis
BackgroundMonoamine oxidase has been implicated in numerous neurological disorders. Although synthetic monoamine oxidase inhibitors (MAOI) have...
