
Abstract
Tauopathies are a heterogenous class of diseases characterized by cellular accumulation of aggregated tau and include diseases such as Alzheimer’s disease (AD), progressive supranuclear palsy and chronic traumatic encephalopathy. Tau pathology is strongly linked to neurodegeneration and clinical symptoms in tauopathy patients. Furthermore, synapse loss is an early pathological event in tauopathies and is the strongest correlate of cognitive decline. Tau pathology is additionally associated with chronic neuroinflammatory processes, such as reactive microglia, astrocytes, and increased levels of pro-inflammatory molecules (e.g. complement proteins, cytokines). Recent studies show that as the principal immune cells of the brain, microglia play a particularly important role in the initiation and progression of tau pathology and associated neurodegeneration. Furthermore, AD risk genes such as Triggering receptor expressed on myeloid cells 2 (TREM2) and Apolipoprotein E (APOE) are enriched in the innate immune system and modulate the neuroinflammatory response of microglia to tau pathology. Microglia can play an active role in synaptic dysfunction by abnormally phagocytosing synaptic compartments of neurons with tau pathology. Furthermore, microglia are involved in synaptic spreading of tau – a process which is thought to underlie the progressive nature of tau pathology propagation through the brain. Spreading of pathological tau is also the predominant target for tau-based immunotherapy. Active tau vaccines, therapeutic tau antibodies and other approaches targeting the immune system are actively explored as treatment options for AD and other tauopathies. This review describes the role of microglia in the pathobiology of tauopathies and the mechanism of action of potential therapeutics targeting the immune system in tauopathies.
Similar content being viewed by others

Introduction
The role of microglia in tauopathies
Pathological tau protein is observed a wide range of neurodegenerative disorders (NDD) and is the key defining feature of a heterogeneous class of diseases called tauopathies. Alzheimer’s disease (AD) is the most common tauopathy - affecting approximately 45 million people worldwide – and is additionally characterized by extracellular plaques composed of amyloid beta (Aβ) [265]. Less common tauopathies include Picks’ disease (PiD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain disease (AGD), and chronic traumatic encephalopathy (CTE). In AD and other tauopathies, tau pathology closely correlates with neurodegeneration and functional decline [11, 115, 147, 211, 232]. Additionally, tauopathies are characterized by early synaptic dysfunction. Tau-induced damage in synaptic compartments ultimately leads to major synapse loss, which is the closest correlate of cognitive decline [76, 148, 263, 264]. Furthermore, synaptic connections are the principal sites at which pathological tau can spread from diseased to healthy neurons – a process which is thought to underlie the progressive nature of tau pathology throughout the brain [218]. Tauopathies are also characterized by reactive gliosis and an increase in inflammatory molecules such complement proteins and pro-inflammatory cytokines – collectively referred to as neuroinflammation [65, 79, 106, 144, 205, 233, 244, 267, 271, 272, 274]. The purpose of the neuroinflammatory state is to remove the cause (e.g. pathogens, protein aggregates, damaged cells) and return the tissue to homeostasis. However, it is not clear if neuroinflammation in tauopathies is mostly protective or damaging and how this depends on disease stage.
Multiple cell types can have immune functions in the brain, for example microglia, astrocytes, perivascular macrophages, meningeal macrophages, choroid plexus macrophages, and infiltrating peripheral myeloid cell types [304]. However, microglia are of particular interest as they are the principal macrophages of the CNS and exciting recent research has shown novel roles for these immune cells in both health and disease. Additionally, genome-wide association studies (GWAS) have identified several late onset AD (LOAD) risk variants that are found in proteins that are predominantly expressed in the innate immune system and microglia (e.g. APOE, TREM2, ABCA7, CD33, CR1) [207]. This strongly implicates microglia as central players in the development of LOAD [124]. Given the central role of tau pathology in AD and other tauopathies, there is now increasing interest in how microglia are involved in the pathobiology of tau protein. It is currently unclear if altered microglial function is a cause, consequence, or contributor to tau pathology. Secreted factors from microglia may lead to initiation of tau aggregation in neurons [116]. Microglia may be also involved in tau-induced synapse loss and tau spreading, and play an important role in the mechanism of action of tau immunotherapy and other therapeutics aimed at treating tauopathies [12, 99, 135, 192]. This review provides an overview of how interaction of tau pathology and microglia leads to synaptic dysfunction in tauopathies. Furthermore, we provide an overview of the published preclinical in vivo studies of tau immunotherapy and immune-related pathways for the treatment of tauopathies.
Tau pathology
Tau is an abundant protein that is predominantly expressed in the axonal compartment of neurons, but also at lower levels in oligodendrocytes and astrocytes [16]. The main function of tau is to regulate the assembly, nucleation and bundling of microtubules and to modulate axonal transport [122]. In addition, recent research suggests that tau may also have a multitude of other physiological functions [282]. Tau protein is encoded by the microtubule-associated protein tau (MAPT) gene on chromosome 17q21.31, and this gene can be mutated, inverted, duplicated, and abnormally methylated. All these modifications have been associated with increased risk of develo** tauopathy and the genetic evidence therefore clearly links tau to neurodegeneration [18, 138, 139, 176].
The human brain contains 6 isoforms generated by alternative splicing of exons 2, 3 and 10 of the MAPT gene [314]. Tau can have either 0, 1 or 2 N-terminal inserts and either 3 or 4 pseudo-repeats (R), resulting in isoforms ranging from 352 to 441 amino acids (aa) (36.7–45.9 kDa) (Fig. 1) [112]. Tau protein can be subdivided into several domains: a structurally disordered N-terminal, the proline rich mid-domain and a highly conserved C-terminal which includes microtubule binding repeats (MTBR). Tau is also subject to a wide range of post-translational modifications (PTMs) (e.g. phosphorylation, acetylation, truncation), which alter its structure, function, and subcellular localization [171, 325]. The six isoforms in combination with the multitude of potential PTMs make the biology of tau extraordinarily complex. Belonging to the class of natively unfolded or intrinsically disordered proteins, tau proteins lack clearly defined secondary and tertiary structures.

Major tau domains and phosphorylation sites. The amino acid sequence of the longest isoform of tau protein (2N4R, 1–441aa) in the central nervous system can be roughly divided into the projection domain on the N-terminal and the microtubule assembly domain on the C-terminal half of the protein. Tau can have up to two inserts in the N-terminal (here shown as N1, N2) and three or four repeats on the C-terminal (R1, R2, R3, R4). These combinations lead to a total of six different isoforms in the central nervous system. The VQIVYK sequence in R2 and VQIINK sequence in R3 are important for aggregation of tau. Several important phosphorylation sites that are associated with tau pathology are shown (p202/205, p212/214, p231, p396/404). These sites are targets for widely used antibodies such as AT8 or PHF1. Several C-terminal truncations have been identified that promote aggregation. Two wellcharacterized truncations are shown here (Δ391 and Δ421)
The MTBR of tau contains two hexapeptides that can form intermolecular beta sheet rich structures: aa275–280 (VCIINK) in R2 and aa306–311 (VQIVYK) in R3 [307, 308]. Pathological conformations of tau can interact with physiological tau, leading to aggregation and ultimately formation of highly structured insoluble fibrils which deposit into the cell as neurofibrillary tangles (NFTs). This process is referred to as templated misfolding, seeded nucleation, or simply seeding [97]. As tau is a highly soluble protein and the initial aggregation phase is thermodynamically unfavorable, it is currently unclear how tau shifts from its dynamic physiological structure to a misfolded monomer that is prone to aggregation [212, 270]. Specific patterns of PTMs may change the conformation of the protein, causing tau to become seed-competent [62, 77]. Moreover, dynamic phosphorylation of the residues in the MTBR or flanking regions regulates the affinity of tau for tubulin and hyperphosphorylation may thereby increase the pool of free tau available for aggregation [160]. The phosphorylation of tau is regulated by both kinases (e.g. cdk5, GSK-3β, p38-MAPK) and phosphatases (e.g. PP2A) [142]. Phosphorylation at a number of sites on tau has been linked to tau pathology (e.g. Ser202/Thr205, Thr212/Ser214, Thr231, Ser396/Ser404, Fig. 1) [314]. Abnormal cleavage can potentially play an important role in tauopathies, as several truncated fragments have an increased propensity for aggregation and their overexpression leads to neurofibrillary pathology in rodents [87, 329]. As will be discussed later in this review, factors secreted from microglia can lead to abnormal patterns of PTMs and may therefore play a role in the initiation of tau aggregation.
Smaller tau oligomers are still soluble and can mislocalize to the somatodendritic compartment to cause toxicity throughout the cell [325]. For this reason, intracellular tau oligomers are also the most toxic species for synapses [120]. In addition to causing intracellular toxicity, tau oligomers and short fibrils can be secreted into the extracellular space and taken up by healthy neurons [98, 121, 162]. This process may be of critical importance as it is thought to underlie the progression of tau pathology throughout the brain. Interestingly, it has already been observed in the classical Braak staging scheme that the progression of tau pathology seems to occur along neuronal connections [43]. It has been demonstrated using a variety in vitro and in vivo approaches that tau pathology predominantly spreads along synaptic connections [48, 73, 318]. Recent studies have made significant progress in showing that this also occurs in the brain of Alzheimer’s patients: seed-competent tau is present in axons of white matter tracts and synaptosomes, and tau seeding occurs in synaptically connected areas before the occurrence of hyperphosphorylated tau in these regions [78, 100, 158, 159]. It is currently unclear what the major mechanism of synaptic tau secretion is, but the evidence so far suggests: (1) release from synaptic vesicles [242] (2) secretion in extracellular vesicles such as exosomes [241, 256, 313] and ectosomes [80], (3) direct translocation across the membrane [157, 208] or (4) tunneling nanotubes [1, 295]. Similarly, several tau uptake mechanisms have been identified which are not mutually exclusive: (1) bulk endocytosis [98, 121, 259, 317] macropinocytosis by heparin sulfate proteoglycans [84, 132, 162, 248, 288, 328] or (3) clathrin-mediated endocytosis [49, 82]. After tau seeds enter the neuron they can seed physiological monomers, thereby propagating the disease process [85].
Neuronal stress or neuronal damage induced by intracellular tau pathology can also impact nearby immune cells, such as microglia [174]. Furthermore, microglia can be affected by extracellular tau secreted by neurons with tau pathology and tau filaments leaking from dying cells [257]. Microglia may also be directly involved in tau-induced synapse loss and synaptic spreading of tau pathology [12, 75]. Understanding how microglia contribute to synaptic dysfunction is therefore of critical importance and will be discussed in more detail below.
Microglia and their role at the synapse
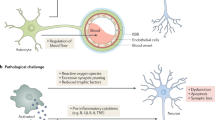
Microglia are the tissue resident macrophages in the brain and originate from yolk-sac-derived erythro-myeloid progenitors [113]. Their unique identity - which distinguishes them from other macrophages in the brain - is the result of this ontogeny and the characteristic micro-environment in the brain [25]. Once microglia have established themselves in the brain during development, their colony is maintained through continuous self-renewal [3, 213]. Furthermore, developmental pruning by microglial phagocytosis is critical for normal brain development [234, 262]. Knockout of the chemokine (C-X3-C motif) ligand 1 (Cx3cl1) receptor leads to reduced microglial synaptic pruning, altered synaptic function, neural connectivity, and social behavior [39, 136, 234, 326]. It is unclear how loss of CX3C chemokine receptor 1 (Cx3cr1) leads to pruning deficits, but it is possible that the chemokine Cx3cl1 acts as a soluble “find-me” signal for microglia. In addition, P2Y12 purinergic receptors may also act as receptors that respond to “find me” signals from synapses. P2Y12 receptors are required for process outgrowth to damaged tissue [125, 193] and also modulate synaptic plasticity in visual cortex [280]. A more direct pathway is a surprising new role for the complement system. Complement initiation factor C1q tags synapses for removal in an activity dependent manner. This subsequently leads to deposition of complement component 3 (C3) and microglial phagocytosis via complement receptor 3 (C3R) [262]. This pathway seems to be reactivated under neurodegenerative conditions and this will be discussed in later sections of this review. A comprehensive understanding of the signals that lead to localization of C1q at synapses is still missing, but it is known that microglia are the dominant source of C1q [92]. Additionally, astrocytic TGF-β signaling can induce C1q expression in develo** retinal neurons and blocking this pathways blocks synapse removal [31]. Astrocytes also secrete interleukin-33, which acts on microglial interleukin 1 receptor-like 1 to promote synapse phagocytosis [302]. Microglial synapse phagocytosis via triggering receptor expressed on myeloid cells 2 (TREM2) – which is encoded by a LOAD risk gene - also plays a role in normal development of neural circuits [88]. Developmental synaptic pruning by microglia is a tightly regulated process as microglia also respond to “don‘t eat me” signals such as cluster of differentiation 47 (CD47) to prevent excess pruning [180].
Microglia also play an important role in maintaining synaptic structure and function later in life. Microglia are for example required for maintenance of synaptic structure and synaptic transmission in the adult retina [312]. Microglia-synapse contacts were also shown to enhance synaptic activity and promote neuronal network synchronization [4]. Furthermore, activated microglia can protect the adult brain by migrating towards inhibitory synapses and displacing them from cortical neurons [55]. Interestingly, microglia play a role in the adult brain by learning dependent synapse formation via secretion of brain-derived neurotrophic factor (BDNF) [235]. Microglial cytokines interleukin (IL)-1 beta (1B), IL-2, IL-6, IL-8, IL18, interferon (IFN)-alpha, INF-gamma and tumor necrosis factor alpha (TNF-a) are all involved in synaptic plasticity, learning, and memory [225]. Low levels of even pro-inflammatory cytokines might therefore be necessary for normal synaptic function. Microglia thus have important physiological functions at the synapse in both the develo** and adult brain.
Microglia in the aging brain
When trying to understand the effects of pathological protein aggregates such as tau pathology on the brain, it is important to note that in humans these effects are often superimposed on the normal effects of aging. In rats, for example, viral delivery of tau protein to young and aged animals led to more microgliosis, neuronal loss, and behavioral deficits in the aged group [166]. It is therefore also important to understand the normal alterations of microglia in the aging brain. For example, a somatic mutation in microglia precursor cells leads to late-onset neurodegeneration [201], which suggests that genetic phenotypes of microglia can manifest themselves in the context of the aging. It is therefore possible that the effects of late onset AD risk mutations in proteins expressed in microglia only become apparent at advanced age. Indeed, haploinsufficiency of AD risk gene TREM2 only leads to impaired response of microglia to injury in old mice [261]. Furthermore, in old age, microglia operate in an aged environment. For example, age-related myelin fragmentation overloads the microglial lysosomal system and contributes to microglial senescence and immune dysfunction in aging [255].
Microglia not only respond to the aging cells around them but also display signs of senescence themselves in the aging human brain [231, 292]. In vivo imaging of young, adult, and very old mice shows changes in morphology and behavior in addition to a slight increase in cell density [126]. The transcriptional microglial phenotype in aging and chronic neurodegeneration is different from acute microglial activation by lipopolysaccharide (LPS) [133]. Microglial genes that encode proteins involved in the scanning of the brain parenchyma - the so-called ‘sensome’ – change their expression in aging [130]. Although mouse and human microglia have a large overlap in expression patterns, these genetic networks start to diverge in aging [101, 117]. The genes that are different in aging are associated with actin dynamics and the sensome, indicating that mouse and human microglia age quite differently. Furthermore, microglia also show regional variation in gene expression in aging, indicating that some brain regions may be more vulnerable to aging of the innate immune system [118]. Taken together, microglial senescence may impair their ability to keep the aging brain clean.
Bidirectional effects of tau pathology and microglial neuroinflammation
The effects of tau pathology on microglia
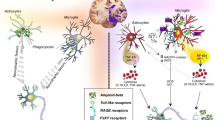
In AD, microglia were previously predominantly studied in the context of plaque pathology and plaque-associated microglia were indeed already observed by Alois Alzheimer [7]. However, reactive microglia, reactive astrocytes, and inflammation-associated molecules are also observed around neurofibrillary tangles (NFTs) and ghost NFTs in AD brains [65, 79, 119, 233, 244, 269, 271, 274]. Furthermore, the same is also observed in primary tauopathies such as PiD, CBD, PSP, Guam Parkinson, Anti-IgLON5 disease [21, 56, 105, 106, 127, 128, 144, 237, 267], and tau transgenic animals [13, 140, 260, 289, 322, 331, 332]. As will be described in more detail later, tau pathology is also robustly associated with activation of classical complement cascade and the release of pro-inflammatory cytokines such as IL1B, IL6 and TNFa [182]. A variety of factors can potentially mediate tau-induced neuroinflammation (Fig. 2a).

a Several cell types are involved in tau-induced neuroinflammation. Neurons with tau pathology exposing phosphatidylserines can be live phagocytosed by microglia. Neuronal tau pathology also induces neuroinflammation by shedding myelin fragments, secreting stress factors, tau oligomers, or via other unknown pathways. In Alzheimer’s disease – the most common tauopathy – extracellular amyloid plaques also induce neuroinflammation. Tau oligomers can damage the vasculature directly, or indirectly via microglia-induced neuroinflammation or alterations of astrocytic functions at the vasculature. All these events can potentially lead to exacerbation of the neuroinflammatory state, which in turn can aggravate tau pathology via proinflammatory cytokines. Microglia can also induce a neurotoxic “A1” phenotype in astrocytes which directly leads to neurodegeneration. Astrocytes in primary tauopathies can also accumulate tau, which can lead to mild changes in the vasculature and possibly impact microglia and synaptic function. b Microglia and astrocytes play an important role in tau-induced synaptic dysfunction. Microglia can phagocytose synapses from neurons with tau pathology via the classical complement pathway. Microglia can also phagocytose secreted tau oligomers and spread them to healthy neuron in exosomes. Microglia in the healthy brain also play an important role in synapse homeostasis, for example via the secretion of cytokines or secretion of growth factors. Tau pathology could alter these homeostatic functions and lead to possible toxic gain-of-function. Astrocytes also play a critical role in synaptic function, for example by taking up extracellular glutamate, release of gliotransmitters that act on synaptic receptors, and secretion of factors that promote synapse assembly. Microglia in tauopathies can also alter the homeostatic functions of astrocytes, possibly leading to synaptic toxicity. Astrocytes with tau pathology can potentially also have deleterious effects on synaptic functions, but this is not yet studied and the role of microglia is therefore unclear
The most obvious one is that tau aggregates directly activate microglia. Tau oligomers co-localize with microglia, astrocytes, and pro-inflammatory cytokines in the brains of tauopathy patients and transgenic mice [222]. When applied in vitro, tau monomers, oligomers, and fibrils directly cause alterations in microglial morphology and secretion of pro-inflammatory cytokines [216, 238]. Microglia have the capacity to phagocytose tau aggregates in vitro and in vivo [12, 44, 73, 74, 99, 135, 192] and recent research shows that the process is partly dependent on Cx3cr1 receptors [37, 38]. Although the phagocytic capacity of microglia for tau aggregates seems to be relatively modest [135, 196], tau aggregates are consistently found in reactive microglia in patient brains [135, 229]. It is unclear if tau aggregates cause microglial activation after phagocytosis or if they are recognized by microglial surface receptors leading to pro-inflammatory cytokine release. Overexpression of full-length tau in microglia causes their activation, but it is unclear how this finding relates to the uptake of tau by microglia in the brain [310].
Interestingly, application of AD-derived soluble tau to cultured microglia causes their degeneration [251, 257] and dystrophic microglia in the aged marmoset often contain hyperphosphorylated tau [249]. Indeed, during aging and AD, altered cytoskeleton, morphology, and senescence of microglia have stronger correlation with tau pathology than microglial activation [17, 72, 249, 291, 292, 297]. It is therefore possible that microglia first have the ability to phagocytose extracellular tau, but they are ultimately not able to keep up with degrading insoluble material around them. This leads them to become dystrophic and lose their normal homeostatic functions [135, 290]. Microglia also show regional variation in clearance of dying neurons and dysfunctional synapses, which may contribute to regional vulnerability to tauopathy [15]. Neuronal tau pathology leads to accumulation of senescent microglia and astrocytes and removal of these senescent cells from a mouse model of neuronal tauopathy led to decreased tau pathology and improved cognition [47]. The presence of dysfunctional glial cells can thus directly contribute to neuronal tau pathology.
Neurons that are trying to cope with tau pathology express factors such as Cx3Cl1 acting on microglial receptor Cx3cr1 [174]. This signaling mechanism limits overactivation of microglia, and these types of pathways are therefore referred to as immune checkpoints [129]. In aged mice or animal models with Aβ plaque deposition, receptors for immune checkpoints are downregulated in microglia [161]. Once tau-induced degeneration of the neuron progresses, intracellular components, myelin debris, and intracellular tau aggregates may activate microglia. Live neurons with tau filaments expose phosphatidylserines, which act as an “eat-me” signal to microglia. Microglia then secrete the opsonin milk-fat-globule EGF-factor-8 and nitric oxide, leading to live phagocytosis of the neuron [44]. It is currently unclear, however, if this is process is harmful or helpful. Microglial phagocytosis of stressed-but-viable neurons may lead to cognitive decline via disintegration of neuronal networks [46]. On the other hand, preventing phagocytosis of live neurons or neuronal compartments with tau filaments may cause inflammation and leakage of aggregated tau which can spread to healthy neurons [293].
The effects of tau pathology on microglia may also be mediated by the vasculature. Tau pathology leads to vascular inflammation and alterations of blood vessels [27, 151, 163, 194, 210], which may be caused by accumulation of tau oligomers and fibrils in the microvasculature [34, 52, 210]. Indeed, early AD is already associated with cerebrospinal fluid (CSF) markers of cerebrovascular inflammation which is associated with phosphorylated tau [149]. Additionally, tau-induced neuroinflammation can damage the blood brain barrier (BBB) which may in turn exacerbate inflammation as blood components activate microglia [71, 170]. Furthermore, tau pathology was reported to lead to infiltration of peripheral immune cells, via secretion of microglial chemokine CCL3 and increased expression of endothelial signaling molecules [175, 195]. This process was associated with neuroinflammation and depleting peripheral T-cells with an anti-CD3 antibody reduced tau pathology-induced expression of pro-inflammatory cytokines and cognitive deficits. Microglia may therefore adversely affect the vasculature downstream of tau pathology, but could in turn also get affected by vascular abnormalities and alterations in the BBB. Interestingly, astrocytes also contain tau inclusions in primary tauopathies such as PSP, CBD, PiD, as well as in the aging brain [131]. A mouse model of astrocytic tau pathology contains tau inclusions in the astrocytic endfeet associated with vasculature. This is accompanied by accumulation of IgG and albumin around the blood vessels, indicative of mild BBB disruption that may in turn lead to microglial activation [95].
Microglia contribute to tau pathology
Whether inflammation is a cause, a contributor, or a consequence of tau pathology is one of the central questions relating to the role of microglia in tauopathies [330]. Several studies have used genetic approaches in mice to examine the relationship between microglia, inflammation, and tau pathology. As mentioned previously, Cx3cl1 acts on microglial receptor Cx3cr1 to limit microglia-induced neuroinflammation. Cx3cl1 overexpression in a tauopathy mouse model decreases tau hyperphosphorylation of tau, neurodegeneration, and cognitive deficits – likely by suppressing microglial activation via Cx3cr1 [89, 221]. The opposite effect was observed in CX3CR1 receptor knockouts [22, 29, 178, 199]. Curiously, knockout of tau also rescued inflammation-mediated neurodegeneration in mice lacking the Cx3cr1 receptor [198]. This indicates that endogenous tau may protect against inflammation and its downstream effects via a yet-unknown mechanism. The deletion of small GTPase RhoA specifically from microglia led to microglial activation, astrogliosis, increased transcription levels of pro-inflammatory cytokines, neurodegeneration, and accumulation of hyperphosphorylated tau in wild-type mice [281]. This suggests that microglia-induced inflammation could not only aggravate existing tau pathology, but potentially also initiate accumulation of hyperphosphorylated tau. In addition to the effects of specific genes, transgenic animals bred on a background that is more prone to neuroinflammation have increased neurofibrillary pathology, despite similar expression levels of truncated tau [289]. These studies show that inflammation can directly lead to initiation or aggravation of tau pathology and its associated consequences.
Administration of anti- or proinflammatory stimuli or compounds has been used to demonstrate that microglia and inflammation are linked to tau pathology. Treatment of mouse models of tauopathy with anti-inflammatory drugs led to a decrease in tau pathology [104, 322]. Furthermore, depletion of microglia with drugs that block colony-stimulating factor-1, which is critical for microglial survival, led to a decrease in accumulation of hyperphosphorylated tau in a mouse model of tauopathy [12], but not in the 3xTG mouse model that develops both tau pathology and Aβ plaques [68]. Additionally, reduction of microglia using the same approach in an aged aggressive tauopathy model did not lead to changes in tau pathology or neurodegeneration [26]. Approaches to reduce inflammation in mouse models in tauopathy will be described in more detail in later sections of this review and are summarized in Table 1.
Induction or exacerbation of inflammation is also likely linked to tau pathology. Both administration of LPS and virus-induced inflammation led to increased hyperphosphorylated and insoluble tau in 3xTg mice and this effect could be rescued by blocking the kinase GSK-3B [165, 294]. LPS also accelerates accumulation of hyperphosphorylated tau in the aggressive rTg4510 tauopathy model, but this was not associated with more Gallyas-positve NFTs [177]. Importantly, LPS was even shown to induce accumulation of phosphorylated tau in wild-type mice [102, 250]. In addition, administration of viral mimic polyriboinosinic-polyribocytidilic acid also led to peripheral inflammation, release of pro-inflammatory cytokines, missorting of tau to the somatodendritic compartment, and accumulation of hyperphosphorylated tau in wild-type mice [173]. Thus, neuroinflammation could not only exacerbate ongoing tau pathology but potentially also lead to the earliest pathological events of tau pathology. How microglial inflammation might worsen or even possibly initiate tau pathology is an important question, which will be the topic of the next sections.
The role of the complement pathway in tau pathology
One consistently upregulated pathway in tauopathies is complement [305]. The complement system is part of the innate immune system and enhances the ability of antibodies and phagocytes to clear pathogens and damaged cells. Complement consists of three potential initiating pathways that all converge on the formation of a C3 convertase which cleaves C3 into C3a and C3b [305], which then cleaves C5 into C5a and C5b. C3a and C5a are anaphylatoxins that play an important role in attracting immune cells and increasing inflammation [305]. C3b on the other hand binds to pathogens or damaged cells and interacts with C3R on phagocytes such as microglia to enhance phagocytosis [305]. C5b plays an import role in the membrane-attack-complex (MAC). The MAC disrupts the integrity of the cell membrane and leads to death and lysis of the cell [305].
The complement pathway has been studied at multiple levels in the context of tau pathology. For example, overexpression of natural C3 inhibitor sCrry was found to decrease tau pathology [45]. Accordingly, knocking out C3aR - the receptor for the chemo-attractant peptide C3a (C3aR) – led to the rescue of hyperphosphorylated and misfolded tau accumulation [190]. The tauopathy mice without the C3aR also had almost no signs of neuroinflammation, synapse loss, neurodegeneration, and cognitive deficits [190]. More downstream components of the complement pathway are most likely also involved in tauopathy. C5a receptors (e.g. C5aR) were shown to be closely associated with NFTs in human brains [93] and C5aR antagonists decrease tau pathology in 3xTG-AD [90]. Proteins of the MAC are also located on neurons with NFTs [146, 206, 287, 315, 324] and an increase in MAC formation was shown to lead to increased tau pathology and neuron loss [45]. Curiously, however, knocking out C1q - the initiating factor of the classical complement pathway – had no effect on neuroinflammation and tau pathology in the 3xTG-AD mouse model [91]. Collectively, these results show that the multiple parts of the complement pathway regulate tau accumulation and its downstream consequences.
The role of microglial secreted factors in tau pathology
The mechanism of inflammation-induced tau pathology seems to be at least partly mediated through the direct effect of pro-inflammatory cytokines. The best characterized cytokine involved in this regard is IL1B, which is cleaved into its active form by caspase 1 – downstream of NLRP3 inflammasome activation [276]. Indeed, the inflammasome is robustly upregulated in response to aggregated tau [284]. IL1B increased the accumulation of hyperphosphorylated tau and was associated with reductions in synaptic marker synaptophysin in vitro [169, 185]. This effect was replicated in vivo and a number of studies have now shown using a variety of genetic and pharmacological approaches that this effect was mediated via the inflammasome and ultimately leads to hyperphosphorylation of tau by the kinases cdk5/p25, GSK-3β and p38-MAPK [29, 57, 107, 164, 197, 199, 273]. The cytokine IL-18 is also a product of the NLRP3 inflammasome and was shown to induce kinases that led to tau hyperphosphorylation [230]. The strongest evidence for inflammation-induced initiation of tau pathology currently exists for TNFa. This cytokine is almost exclusively expressed in microglia and can cause formation of tau aggregates in neuronal neurites in vitro via the formation of reactive oxygen species [116]. Furthermore, overexpression of TNFa in 3xTG-AD mice led to increased tau pathology [150]. Knockout of TNF-R2 or both TNF-R1 and TNF-R2 in the same mouse model led to increased plaque and tau pathology [214, 215]. It is therefore possible that both TNFa receptors have complex and opposing effects on the development of tau pathology, but more studies in mouse models of pure tauopathies are needed. As mentioned previously, the cytokine IL6 is also consistently upregulated in tauopathy mouse models. IL6 leads to phosphorylation of tau at AD-associated residues via deregulation of the cdk5/p35 pathway [245]. In addition to the effects of cytokines on kinases and phosphates, it was recently shown that metalloproteinase MMP-9 causes tau aggregation via deacetylase HDAC6 [The role of astrocytes in tau-induced synaptic dysfunction Microglia also have bidirectional signaling cascades with astrocytes. Astrocytes are a highly heterogenous population that make up approximately 20% of brains cells and are derived from the same progenitors as neurons [6]. Astrocytes have a wide range of functions, including providing nutrient support to neurons, forming part of the BBB, and modulating the flow of CSF in the brain as part of the glymphatic system [6, 247]. Astrocytes have highly ramified processes and it is estimated that a single cortical astrocyte can contact up to 100,000 synapses in mice and up to 2,000,000 synapses in humans [5]. Indeed, astrocytes play a critical role in neuronal connections by regulating glutamate homeostasis, secreting gliotransmitters (e.g. ATP), secreting factors that promote assembly and plasticity of synapses (e.g. thrombospondins), and synaptic phagocytosis (e.g. via MERTK and MEGF10) [6]. Under a variety of disease and neurodegenerative conditions, microglial cytokines (IL1a, TNFa and C1q) can induce a unique transcriptional profile in astrocytes that is characterized by dramatic upregulation of complement protein C3. This was associated with a neurotoxic phenotype termed “A1 astrocytes”, characterized by secretion of neurotoxic factors, loss of neurotrophic functions, and impairments in several homeostatic synaptic functions [188]. A1 astrocytes can be induced in normal aging mice and are associated with more severe neurodegeneration in a mouse model of tauopathy [36, 61, 277]. Interestingly, microglia also secrete factors (e.g. TGFα, VEGF-B) that limit the pathogenic activities of astrocytes [253]. Furthermore, C3 upregulation in astrocytes is not only the result of microglial inflammation, but the downstream cleavage product C3a can in turn dramatically increase the synaptic toxicity of microglia in mouse models of amyloidosis and tauopathy by binding to microglial C3aRs [186, 187, 190]. Cross-signaling between microglia and astrocytes therefore plays a key role in modulating synaptic dysfunction and neurodegeneration (Fig. 2b). Tau pathology can lead to synapse loss via a decrease in neurotrophic thrombospondin signaling by astrocytes [278]. In addition, impaired gliotransmitter release from astrocytes was also shown to mediate tau-induced synaptic dysfunction [239]. It is expected that neurofibrillary pathology-induced loss of astrocytic glutamate homeostasis causes neuronal network dysfunction and potential excitotoxicity. However, an interesting study shows that healthy subjects with NFTs had more activated astrocytes with increased glutamate transporter 1 expression compared to AD cases with dementia [167]. This raises the possibility that at least some astrocytic phenotypes observed in tauopathies may be beneficial rather than damaging. Astrocytes can also prune synapses in the healthy brain [59, 60] or under disease conditions, such as ischemia [217] and sleep deprivation [19]. Furthermore, astrocytes were shown to phagocytose apoptotic cells via the C1q-MEGF10 pathway [143]. This raises the possibility that not only microglia, but also astrocytes can use the classical complement pathway to phagocytose synapses on living neurons. Astrocytes were also shown to clear dystrophic neurites in a mouse model of AD [114]. Since dystrophic neurites in AD patients often contain aggregated tau, it is possible that astrocytes phagocytose pathological tau species. Furthermore, the close proximity of astrocytes to the pre- and postsynaptic compartments also raises the possibility that astrocytes can pick up secreted extracellular tau or digest damaged synapses with hyperphosphorylated tau [73]. Intriguingly, recent studies show that astrocytes may also be involved in tau spreading along neuronal connections or from astrocyte-to-astrocyte [200, 220]. Finally, a transgenic mouse model of astrocytic tau pathology displayed reduced expression and function of glutamate transporter-1, and motor impairments already before disease stages with overt hyperphosphorylated tau accumulation [67]. This indicates that astrocytic tau pathology may lead to alterations in synaptic glutamate homeostasis, neuronal network dysfunction, and associated functional impairments .
The role of microglia in therapeutic approaches targeting the immune system in tauopathies
Given the increasing recognition of microglia as central players in the pathogenesis of tauopathies, it is perhaps not surprising that there is increasing interest in targeting inflammatory pathways for these diseases (Table 1). Anti-inflammatory compounds such as FK506 and minocycline were shown to reduce tau pathology and downstream neurodegeneration, but their mechanism of action in relation to tau pathology is unclear [104, 226, 322]. Depletion of immune cells such as microglia or T-cells may also be efficacious when initiated at early stages of tau pathology [12, 175]. However, it is important to keep in mind that the immune system in the periphery and the brain plays an important physiological role. Non-specific suppression of the immune system could leave the patient vulnerable to increased risk of infections and accumulation of cellular debris in the context of neurodegeneration. Targeted pharmacological removal of dysfunctional cells from the brain may in itself be an efficacious therapeutic approach for tauopathies [47]. Furthermore, restoring homeostatic microglial phagocytosis with a CD22-blocking antibody rescued cognitive deficits in aged mice, indicating that normal microglial function is critical for maintaining a normal cognitive state [240]. There is therefore increasing focus on targeting specific inflammatory targets such as complement [75, 190], leukotrienes [108, 110], or the NLRP3 inflammasome [284]. Antibodies against C1q can directly reduce tau pathology-induced synapse phagocytosis by microglia [75]. It remains to be determined to what extent microglial synapse phagocytosis plays a central role in the pathogenesis of tauopathies, and if there are other mechanisms through which microglia affect tau-induced synaptic dysfunction that could be targeted with therapeutic agents.
Immunotherapy directly targeting tau protein has also emerged as a promising approach in the treatment of tauopathies and microglia have been increasingly recognized as an important player in the mechanism of action of immunotherapeutic approaches [228]. Active tau immunotherapy has been pursued in multiple varieties: immunization with full-length tau, peptides corresponding to domains on tau, or peptides that correspond to tau phosphorylated at specific residues. Most of these approaches led to reductions in measures of tau pathology, such as sarkosyl insoluble tau and NFTs (Table 2). As tau vaccines trigger an active immune response, patient safety must be carefully monitored. Although in several pre-clinical tau vaccination studies some adverse effects ranging from paralysis caused by severe neuroinflammation to microgliosis have been described [24, 35, 252, 254, 296], these studies used strong T helper 1 inducing adjuvants, which are not used in humans because of safety concerns [63]. Studies that used milder adjuvants have reported similar efficacy, but either no change or reductions in microgliosis [[9, 30, 42, 152, 246, 268] (summarized in Table 2). Passive immunotherapy with monoclonal antibodies offers the advantage of tight control over antibody binding characteristics and titers in the blood. Antibodies binding to specific domains on tau, specific tau phosphorylation sites, pathological tau conformations, and tau oligomers have been developed. Various tau antibodies can decrease sarkosyl insoluble tau, NFTs, and tau spreading (Table 3). Except for one study (see [69]), no differences in gliosis or pro-inflammatory cytokines have been described in passive immunotherapy studies that tested for neuroinflammation [[41, 50, 51, 53, 54, 66, 258] (summarized in Table 3). However, it should be noted that the majority of studies did not report the effects of passive immunotherapy on microglia or other markers indicative of neuroinflammation.
Most research has focused on the development of antibodies that neutralize extracellular tau and inhibit synaptic spreading of pathological tau [227, 320]. Tau antibodies could neutralize extracellular tau oligomers before they have the chance to damage glial cells and the vasculature. As mentioned previously, both microglia and astrocytes are involved in propagation of tau pathology [12, 200]. Antibody-mediated neutralization of tau seeds before they reach these types of cells, may therefore diminish downstream exosomal tau spreading. Additionally, extracellular tau aggregates may lead to reactive gliosis, which can potentially be inhibited by anti-tau antibodies. Extracellular tau aggregates neutralized by tau antibodies need to be removed from the brain. Clearance of extracellular tau or other macromolecules is mediated by the glymphatic system and impairment of this system worsens tau pathology [[141, 236]. In addition to global clearance, tau-antibody complexes in immunized mice can also be cleared locally by means of opsonization via microglial Fc receptors and degradation in the lysosomes [8, 10, 74, 99, 179, 319]. Effectorless antibodies incapable of actively engaging microglia retained their therapeutic effect [179], indicating that tau antibody complexes can also be cleared via additional pathways in immunized mice. Indeed, peripheral or AAV-mediated delivery of tau scFv (without an Fc domain) seems to be an effective therapeutic approach in tauopathy mice [145, 224, 283, 306]. More research is therefore needed to describe the impact of actively engaging microglia with tau immunotherapeutic approaches.
Concluding remarks & future directions
Microglia are fascinating cells and the large number of excellent recent studies demonstrates increasing recognition of microglia as critical players in the pathobiology of tau protein. It is currently not fully resolved if inflammation is a cause, contributor or consequence of tau pathology. Pro-inflammatory mediators secreted from microglia (e.g. cytokines, complement) can initiate tau pathology and play a critical role in tau-induced neurodegeneration. The strongest evidence for inflammation-induced initiation of tau pathology currently exists for TNFa, as this cytokine was shown to lead to formation of tau aggregates in neurites [116]. More preclinical work, however, is needed to fully characterize the immune pathways involved in tau pathology and efforts should be made to validate them in both AD and primary tauopathy patients. Many studies report the effects of neuroinflammatory processes on tau phosphorylation only. Future studies should also focus on the effects of neuroinflammation on oligomerization or accumulation of insoluble tau aggregates. Furthermore, risk genes for AD or other tauopathies will have to be investigated in multiple mouse models of tauopathy, without plaque pathology as a confounding factor. Since the immune response to tau pathology changes as the disease progresses, future studies should also examine the evolution of neuroinflammatory pathways at multiple stages of the disease. Furthermore, more studies are needed on what events cause the initial neuroinflammation in response to tau pathology and via what pathways.
Microglia are also directly involved in tauopathies as they have been shown to pathologically phagocytose synapses of neurons with tau pathology. Currently, the pathways underlying microglia-mediated synapse loss are not fully characterized and a multitude of potential pathways have been identified in neurodevelopment. Complement-mediated synapse loss via microglial synapse phagocytosis under neurodegenerative conditions is now described in multiple disease models. However, it is not known what causes the binding of C1q to synapses and if this happens indiscriminately or only targets vulnerable synapses. In neurodevelopment, there are signals (e.g. CD47) that protect synapses from microglial pruning. We need to understand better the function of these signals in the normal brain and determine if they are downregulated in response to neurofibrillary pathology. Additionally, since astrocytes play a critical role in both synaptic function and neuroinflammation, more studies are needed on bidirectional microglia-astrocyte signaling in tauopathies.
As principal macrophages of the brain, microglia phagocytose tau and may play a role in spreading tau pathology throughout the brain. Determining the exact contribution of microglia to the disease pathogenesis remains an important topic for future investigations. More studies are needed on the mechanisms of tau internalization in microglia and if this is associated with activation of pro-inflammatory pathways. Furthermore, better identification of intracellular pathways that lead to degradation of tau aggregates in microglia is required. This could help explain why microglia in tauopathies are mostly senescent, inefficient in clearing extracellular tau, and potentially become involved in spreading of tau pathology. The loss of the ability of microglia to keep the extracellular space clean of aggregated proteins may play a critical role in the propagation of protein pathology in the brain.
Finally, the role of microglia in the mechanism of action of tau immunotherapy needs to be explored further. Important topics include a better understanding of the fate of tau-antibody complexes in the extracellular space and the associated roles of various antibody isotypes; a better understanding of microglia-mediated clearance mechanisms and other potential clearance pathways like the glymphatic system; and a better characterization of the downstream consequences of Fc-mediated tau antibody internalization. Pre-clinical studies should also report more on the immunological consequences, beyond the mere presence of microgliosis or astrogliosis. Examples would include measuring secreted immune molecules (pro-inflammatory cytokines, complement) and identifying effects on transcriptional phenotypes in glia, such as the recently identified disease-associated microglia and neurotoxic A1 astrocytes [161, 188]. The field of neuroimmunology is advancing rapidly with insights that revolutionize our thinking about the microglia in the brain. Applying these insights to the study of tau pathology will pave the way towards better understanding and treatment of tauopathies.
Availability of data and materials
Not applicable.
Abbreviations
- AAV:
-
Adeno-associated virus
- AD:
-
Alzheimer’s disease
- AGD:
-
Argyrophilic grain disease
- APOE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid beta
- BDNF:
-
Brain-derived neurotrophic factor
- C:
-
Complement
- C3R:
-
Complement receptor 3
- CBD:
-
Corticobasal degeneration
- CD47:
-
Cluster of differentiation 47
- CFA:
-
Complete Freund adjuvant
- CSF:
-
Cerebrospinal fluid
- CTE:
-
Chronic traumatic encephalopathy
- Cx3cl1:
-
Chemokine (C-X3-C motif) ligand 1
- Cx3cr1:
-
CX3C chemokine receptor 1
- DAM:
-
Disease-associated microglia
- Fc:
-
Fragment crystallizable
- GWAS:
-
Genome-wide association studies
- I.c.v.:
-
Intracerebroventricular
- i.p.:
-
Intraperitoneal
- i.v.:
-
Intravenous
- IFN:
-
Interferon
- IL:
-
Interleukin
- LOAD:
-
Late onset Alzheimer’s disease
- LPS:
-
Lipopolysaccharide
- LTP:
-
Long term potentiation
- MAPT:
-
Microtubule-associated protein tau
- MBT:
-
Mycobacteriumtuberculosis H37Ra
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MTBR:
-
Microtubule binding repeats
- NC:
-
No change
- NFT:
-
Neurofibrillary tangle
- PFF:
-
Preformed fibrils
- PiD:
-
Picks’ disease
- PS/PSEN:
-
Presenilin
- PSP:
-
Progressive supranuclear palsy
- PT:
-
Pertussis toxin
- P-tau:
-
Hyperphosphorylated tau
- PTM:
-
Post-translational modification
- scFv:
-
Single-chain variable fragment
- TNF-a:
-
Tumor necrosis factor alpha
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- V BBB:
-
Blood brain barrier
- VLP:
-
Virus-like particles
References
Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C (2016) Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10:344–351. https://doi.org/10.1080/19336896.2016.1223003
Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ (1996) Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol 138:22–32. https://doi.org/10.1006/exnr.1996.0043
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. https://doi.org/10.1038/nn2014
Akiyoshi R, Wake H, Kato D, Horiuchi H, Ono R, Ikegami A, Haruwaka K, Omori T, Tachibana Y, Moorhouse AJ, Nabekura J (2018) Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro 5. https://doi.org/10.1523/ENEURO.0088-18.2018
Allen NJ, Eroglu C (2017) Cell Biology of Astrocyte-Synapse Interactions. Neuron 96:697–708. https://doi.org/10.1016/j.neuron.2017.09.056
Allen NJ, Lyons DA (2018) Glia as architects of central nervous system formation and function. Science 362:181–185. https://doi.org/10.1126/science.aat0473
Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschrift fur Psychiatr und Psych Medizin 64:146–148
Andersson CR, Falsig J, Stavenhagen JB, Christensen S, Kartberg F, Rosenqvist N, Finsen B, Pedersen JT (2019) Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcgamma-receptor binding and functional lysosomes. Sci Rep 9:4658. https://doi.org/10.1038/s41598-019-41105-4
Ando K, Kabova A, Stygelbout V, Leroy K, Heraud C, Frederick C, Suain V, Yilmaz Z, Authelet M, Dedecker R, Potier M-C, Duyckaerts C, Brion J-P (2014) Vaccination with Sarkosyl insoluble PHF-tau decrease neurofibrillary tangles formation in aged tau transgenic mouse model: a pilot study. J Alzheimers Dis 40(Suppl 1):S135–S145. https://doi.org/10.3233/JAD-132237
Apetri A, Crespo R, Juraszek J, Pascual G, Janson R, Zhu X, Zhang H, Keogh E, Holland T, Wadia J, Verveen H, Siregar B, Mrosek M, Taggenbrock R, Ameijde J, Inganas H, van Winsen M, Koldijk MH, Zuijdgeest D, Borgers M, Dockx K, Stoop EJM, Yu W, Brinkman-van der Linden EC, Ummenthum K, van Kolen K, Mercken M, Steinbacher S, de Marco D, Hoozemans JJ, Wilson IA, Koudstaal W, Goudsmit J (2018) A common antigenic motif recognized by naturally occurring human VH5–51/VL4–1 anti-tau antibodies with distinct functionalities. Acta Neuropathol Commun 6:43. https://doi.org/10.1186/s40478-018-0543-z
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42:631–639
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. https://doi.org/10.1038/nn.4132
Asai H, Ikezu S, Woodbury ME, Yonemoto GMS, Cui L, Ikezu T (2014) Accelerated neurodegeneration and neuroinflammation in transgenic mice expressing P301L tau mutant and tau-tubulin kinase 1. Am J Pathol 184:808–818. https://doi.org/10.1016/j.ajpath.2013.11.026
Audrain M, Haure-Mirande J-V, Wang M, Kim SH, Fanutza T, Chakrabarty P, Fraser P, St George-Hyslop PH, Golde TE, Blitzer RD, Schadt EE, Zhang B, Ehrlich ME, Gandy S (2018) Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry. https://doi.org/10.1038/s41380-018-0258-3
Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, Sullivan JM, Purushothaman I, Scarpa JR, Goate AM, Busslinger M, Shen L, Losic B, Schaefer A (2018) Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci 21:1049–1060. https://doi.org/10.1038/s41593-018-0192-3
Ayers JI, Giasson BI, Borchelt DR (2017) Prion-like Spreading in Tauopathies. Biol Psychiatry. https://doi.org/10.1016/j.biopsych.2017.04.003
Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, Patel E, Abner EL, Kryscio RJ, Nelson PT (2015) Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3:32. https://doi.org/10.1186/s40478-015-0209-z
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711
Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C (2017) Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J Neurosci 37:5263–5273. https://doi.org/10.1523/JNEUROSCI.3981-16.2017
Belloy ME, Napolioni V, Greicius MD (2019) A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 101:820–838. https://doi.org/10.1016/j.neuron.2019.01.056
Bellucci A, Bugiani O, Ghetti B, Spillantini MG (2011) Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis 8:221–229. https://doi.org/10.1159/000322228
Bemiller SM, Maphis NM, Formica SV, Wilson GN, Miller CM, Xu G, Kokiko-Cochran ON, Kim K-W, Jung S, Cannon JL, Crish SD, Cardona AE, Lamb BT, Bhaskar K (2018) Genetically enhancing the expression of chemokine domain of CX3CL1 fails to prevent tau pathology in mouse models of tauopathy. J Neuroinflammation 15:278. https://doi.org/10.1186/s12974-018-1310-6
Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, Landreth GE, Lamb BT (2017) TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener 12:74. https://doi.org/10.1186/s13024-017-0216-6
Benhamron S, Rozenstein-Tsalkovich L, Nitzan K, Abramsky O, Rosenmann H (2018) Phos-tau peptide immunization of amyloid-tg-mice reduced non-mutant phos-tau pathology, improved cognition and reduced amyloid plaques. Exp Neurol. https://doi.org/10.1016/j.expneurol.2018.02.004
Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, Barres BA (2018) A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron. https://doi.org/10.1016/j.neuron.2018.05.014
Bennett RE, Bryant A, Hu M, Robbins AB, Hopp SC, Hyman BT (2018) Partial reduction of microglia does not affect tau pathology in aged mice. J Neuroinflammation 15:311. https://doi.org/10.1186/s12974-018-1348-5
Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT (2018) Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1710329115
Bertani I, Iori V, Trusel M, Maroso M, Foray C, Mantovani S, Tonini R, Vezzani A, Chiesa R (2017) Inhibition of IL-1beta Signaling Normalizes NMDA-Dependent Neurotransmission and Reduces Seizure Susceptibility in a Mouse Model of Creutzfeldt-Jakob Disease. J Neurosci 37:10278–10289. https://doi.org/10.1523/JNEUROSCI.1301-17.2017
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68:19–31. https://doi.org/10.1016/j.neuron.2010.08.023
Bi M, Ittner A, Ke YD, Gotz J, Ittner LM (2011) Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One 6:e26860. https://doi.org/10.1371/journal.pone.0026860
Bialas AR, Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16:1773–1782. https://doi.org/10.1038/nn.3560
Biber K, Neumann H, Inoue K, Boddeke HWGM (2007) Neuronal “On” and “Off” signals control microglia. Trends Neurosci 30:596–602. https://doi.org/10.1016/j.tins.2007.08.007
Bie B, Wu J, Foss JF, Naguib M (2019) Activation of mGluR1 Mediates C1q-Dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer’s Rodent Models. Mol Neurobiol. https://doi.org/10.1007/s12035-019-1467-8
Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin Y-C, Zamudio F, Hernandez LD, Sabbagh JJ, Selenica M-LB, Dickey CA (2015) Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun 3:8. https://doi.org/10.1186/s40478-015-0186-2
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H (2010) Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol 224:472–485. https://doi.org/10.1016/j.expneurol.2010.05.010
Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ (2018) The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep 22:269–285. https://doi.org/10.1016/j.celrep.2017.12.039
Bolos M, Llorens-Martin M, Jurado-Arjona J, Hernandez F, Rabano A, Avila J (2016) Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J Alzheimers Dis 50:77–87. https://doi.org/10.3233/JAD-150704
Bolos M, Llorens-Martin M, Perea JR, Jurado-Arjona J, Rabano A, Hernandez F, Avila J (2017) Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener 12:59. https://doi.org/10.1186/s13024-017-0200-1
Bolos M, Perea JR, Terreros-Roncal J, Pallas-Bazarra N, Jurado-Arjona J, Avila J, Llorens-Martin M (2017) Absence of microglial CX3CR1 impairs the synaptic integration of adult-born hippocampal granule neurons. Brain Behav Immunss. https://doi.org/10.1016/j.bbi.2017.10.002
Boon BDC, Hoozemans JJM, Lopuhaa B, Eigenhuis KN, Scheltens P, Kamphorst W, Rozemuller AJM, Bouwman FH (2018) Neuroinflammation is increased in the parietal cortex of atypical Alzheimer’s disease. J Neuroinflammation 15:170. https://doi.org/10.1186/s12974-018-1180-y
Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 118:658–667. https://doi.org/10.1111/j.1471-4159.2011.07337.x
Boutajangout A, Quartermain D, Sigurdsson EM (2010) Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci 30:16559–16566. https://doi.org/10.1523/JNEUROSCI.4363-10.2010
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Brelstaff J, Tolkovsky AM, Ghetti B, Goedert M, Spillantini MG (2018) Living Neurons with Tau Filaments Aberrantly Expose Phosphatidylserine and Are Phagocytosed by Microglia. Cell Rep 24:1939–1948.e4. https://doi.org/10.1016/j.celrep.2018.07.072
Britschgi M, Takeda-Uchimura Y, Rockenstein E, Johns H, Masliah E, Wyss-Coray T (2012) Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J Neuroinflammation 9:220. https://doi.org/10.1186/1742-2094-9-220
Brown GC, Neher JJ (2014) Microglial phagocytosis of live neurons. Nat Rev Neurosci 15:209–216. https://doi.org/10.1038/nrn3710
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. https://doi.org/10.1038/s41586-018-0543-y
Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B, de Wit J, Verstreken P, Moechars D (2015) Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep 11:1176–1183. https://doi.org/10.1016/j.celrep.2015.04.043
Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep 17:931–940. https://doi.org/10.1016/j.celrep.2016.09.063
Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Kayed R (2014) Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis 40(Suppl 1):S97–S111. https://doi.org/10.3233/JAD-132477
Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett ADT, Dineley K, Kayed R (2015) Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J Neurosci 35:4857–4868. https://doi.org/10.1523/JNEUROSCI.4989-14.2015
Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, Gerson JE, Sengupta U, Abisambra J, Nelson P, Troncoso J, Ungvari Z, Galvan V, Kayed R (2017) Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis 8:257–266. https://doi.org/10.14336/AD.2017.0112
Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett ADT, Dineley KT, Jackson GR, Kayed R (2014) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34:4260–4272. https://doi.org/10.1523/JNEUROSCI.3192-13.2014
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, Hutton ML, Citron M (2011) Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 286:34457–34467. https://doi.org/10.1074/jbc.M111.229633
Chen Z, Jalabi W, Hu W, Park H-J, Gale JT, Kidd GJ, Bernatowicz R, Gossman ZC, Chen JT, Dutta R, Trapp BD (2014) Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun 5:4486. https://doi.org/10.1038/ncomms5486
Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. https://doi.org/10.1186/s40478-016-0382-8
Cho S-H, Chen JA, Sayed F, Ward ME, Gao F, Nguyen TA, Krabbe G, Sohn PD, Lo I, Minami S, Devidze N, Zhou Y, Coppola G, Gan L (2015) SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1beta. J Neurosci 35:807–818. https://doi.org/10.1523/JNEUROSCI.2939-14.2015
Chu J, Li J-G, Ceballos-Diaz C, Golde T, Pratico D (2013) The influence of 5-lipoxygenase on Alzheimer’s disease-related tau pathology: in vivo and in vitro evidence. Biol Psychiatry 74:321–328. https://doi.org/10.1016/j.biopsych.2012.12.012
Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. https://doi.org/10.1038/nature12776
Chung W-S, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, Holtzman DM, Barres BA (2016) Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A 113:10186–10191. https://doi.org/10.1073/pnas.1609896113
Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA (2018) Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 115(8):E1896–E1905
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VMY (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2:252. https://doi.org/10.1038/ncomms1255
Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14:399–415. https://doi.org/10.1038/s41582-018-0013-z
Costello DA, Keenan K, McManus RM, Falvey A, Lynch MA (2016) The age-related neuroinflammatory environment promotes macrophage activation, which negatively impacts synaptic function. Neurobiol Aging 43:140–148. https://doi.org/10.1016/j.neurobiolaging.2016.04.001
Cras P, Kawai M, Siedlak S, Perry G (1991) Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res 558:312–314
d’Abramo C, Acker CM, Jimenez HT, Davies P (2013) Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One 8:e62402. https://doi.org/10.1371/journal.pone.0062402
Dabir DV, Robinson MB, Swanson E, Zhang B, Trojanowski JQ, Lee VM-Y, Forman MS (2006) Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci 26:644–654. https://doi.org/10.1523/JNEUROSCI.3861-05.2006
Dagher NN, Najafi AR, Kayala KMN, Elmore MRP, White TE, Medeiros R, West BL, Green KN (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation 12:139. https://doi.org/10.1186/s12974-015-0366-9
Dai C-L, Tung YC, Liu F, Gong C-X, Iqbal K (2017) Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther 9:1. https://doi.org/10.1186/s13195-016-0227-5
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758. https://doi.org/10.1038/nn1472
Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227. https://doi.org/10.1038/ncomms2230
Davies DS, Ma J, Jegathees T, Goldsbury C (2017) Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol 27:795–808. https://doi.org/10.1111/bpa.12456
de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73:685–697. https://doi.org/10.1016/j.neuron.2011.11.033
De Marco D, Taggenbrock R, Crespo R, Koudstaal W, Ramsburg E, Apetri A (2018) Cell-based Assay to Study Antibody-mediated Tau Clearance by Microglia. J Vis Exp:e58576. https://doi.org/10.3791/58576
Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, Foreman O, Carano RAD, Chih B, Klumperman J, Bakalarski C, Hanson JE, Sheng M (2018) Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron. https://doi.org/10.1016/j.neuron.2018.10.014
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464. https://doi.org/10.1002/ana.410270502
Despres C, Byrne C, Qi H, Cantrelle F-X, Huvent I, Chambraud B, Baulieu E-E, Jacquot Y, Landrieu I, Lippens G, Smet-Nocca C (2017) Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1708448114
DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, Commins C, Gonzalez JA, Dooley PM, Frosch MP, Hyman BT (2018) Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front Neurosci 12:267. https://doi.org/10.3389/fnins.2018.00267
DiPatre PL, Gelman BB (1997) Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 56:143–149
Dujardin S, Begard S, Caillierez R, Lachaud C, Delattre L, Carrier S, Loyens A, Galas M-C, Bousset L, Melki R, Auregan G, Hantraye P, Brouillet E, Buee L, Colin M (2014) Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One 9:e100760. https://doi.org/10.1371/journal.pone.0100760
Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM (2014) Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci 34:2618–2631. https://doi.org/10.1523/JNEUROSCI.4200-13.2014
Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, Billinton A, Livesey FJ (2018) Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlap** but Distinct Pathways. Cell Rep 22:3612–3624. https://doi.org/10.1016/j.celrep.2018.03.021
Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu L-J (2014) Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci 34:10528–10540. https://doi.org/10.1523/JNEUROSCI.0416-14.2014
Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, Jackson S, O’Neill MJ, Isaacs AM, Hutton ML, Szekeres PG, Goedert M, Bose S (2015) Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem 290:1049–1065. https://doi.org/10.1074/jbc.M114.589309
Falcon B, Noad J, McMahon H, Randow F, Goedert M (2017) Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem. https://doi.org/10.1074/jbc.M117.809293
Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA (2016) Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging 37:19–25. https://doi.org/10.1016/j.neurobiolaging.2015.09.011
Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, Novak M (2012) First transgenic rat model develo** progressive cortical neurofibrillary tangles. Neurobiol Aging 33:1448–1456. https://doi.org/10.1016/j.neurobiolaging.2010.10.015
Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, Matteoli M (2018) The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity. https://doi.org/10.1016/j.immuni.2018.04.016
Finneran DJ, Morgan D, Gordon MN, Nash KR (2018) CNS-Wide over Expression of Fractalkine Improves Cognitive Functioning in a Tauopathy Model. J Neuroimmune Pharmacol. https://doi.org/10.1007/s11481-018-9822-5
Fonseca MI, Ager RR, Chu S-H, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ (2009) Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol 183:1375–1383. https://doi.org/10.4049/jimmunol.0901005
Fonseca MI, Chu S-H, Berci AM, Benoit ME, Peters DG, Kimura Y, Tenner AJ (2011) Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflammation 8:4. https://doi.org/10.1186/1742-2094-8-4
Fonseca MI, Chu S-H, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14:48. https://doi.org/10.1186/s12974-017-0814-9
Fonseca MI, McGuire SO, Counts SE, Tenner AJ (2013) Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation 10:25. https://doi.org/10.1186/1742-2094-10-25
Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M, Fariss RN, Li W, Wong WT (2011) Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6:e15973. https://doi.org/10.1371/journal.pone.0015973
Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM-Y, Trojanowski JQ (2005) Transgenic Mouse Model of Tau Pathology in Astrocytes Leading to Nervous System Degeneration. J Neurosci 25:3539–3550
Franzmeier N, Rubinski A, Neitzel J, Ewers M (2019) The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat Commun 10:1766. https://doi.org/10.1038/s41467-019-09564-5
Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E (1998) A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A 95:15712–15717
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. https://doi.org/10.1074/jbc.M808759200
Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI (2015) Distinct Therapeutic Mechanisms of Tau Antibodies: Promoting Microglial Clearance Versus Blocking Neuronal Uptake. J Biol Chem 290:21652–21662. https://doi.org/10.1074/jbc.M115.657924
Furman JL, Vaquer-Alicea J, White CL 3rd, Cairns NJ, Nelson PT, Diamond MI (2017) Widespread tau seeding activity at early Braak stages. Acta Neuropathol 133:91–100. https://doi.org/10.1007/s00401-016-1644-z
Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Veras MM, Pereira TF, Leite REP, Moller T, Wes PD, Sogayar MC, Laman JD, den Dunnen W, Pasqualucci CA, Oba-Shinjo SM, Boddeke EWGM, Marie SKN, Eggen BJL (2017) Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 20:1162–1171
Gardner LE, White JD, Eimerbrink MJ, Boehm GW, Chumley MJ (2016) Imatinib methanesulfonate reduces hyperphosphorylation of tau following repeated peripheral exposure to lipopolysaccharide. Neuroscience 331:72–77. https://doi.org/10.1016/j.neuroscience.2016.06.007
Garre JM, Silva HM, Lafaille JJ, Yang G (2017) CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-alpha. Nat Med. https://doi.org/10.1038/nm.4340
Garwood CJ, Cooper JD, Hanger DP, Noble W (2010) Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front psychiatry 1:136. https://doi.org/10.3389/fpsyt.2010.00136
Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord 21:89–93. https://doi.org/10.1002/mds.20668
Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K, Brooks DJ (2004) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord 19:1221–1226. https://doi.org/10.1002/mds.20162
Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK (2013) Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33:5053–5064. https://doi.org/10.1523/JNEUROSCI.4361-12.2013
Giannopoulos PF, Chiu J, Pratico D (2018) Antileukotriene therapy by reducing tau phosphorylation improves synaptic integrity and cognition of P301S transgenic mice. Aging Cell:e12759. https://doi.org/10.1111/acel.12759
Giannopoulos PF, Chiu J, Pratico D (2018) Learning Impairments, Memory Deficits, and Neuropathology in Aged Tau Transgenic Mice Are Dependent on Leukotrienes Biosynthesis: Role of the cdk5 Kinase Pathway. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1124-7
Giannopoulos PF, Chu J, Sperow M, Li J-G, Yu WH, Kirby LG, Abood M, Pratico D (2015) Pharmacologic inhibition of 5-lipoxygenase improves memory, rescues synaptic dysfunction, and ameliorates tau pathology in a transgenic model of tauopathy. Biol Psychiatry 78:693–701. https://doi.org/10.1016/j.biopsych.2015.01.015
Giannopoulos PF, Pratico D (2017) Overexpression of 5-Lipoxygenase Worsens the Phenotype of a Mouse Model of Tauopathy. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0817-7
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3:519–526
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald H-R (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551. https://doi.org/10.1038/nature13989
Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nunez-Diaz C, Sanchez-Varo R, Sanchez-Mico MV, Trujillo-Estrada L, Fernandez-Valenzuela JJ, Vizuete M, Comella JX, Galea E, Vitorica J, Gutierrez A (2017) Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia. https://doi.org/10.1002/glia.23270
Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41:17–24. https://doi.org/10.1002/ana.410410106
Gorlovoy P, Larionov S, Pham TTH, Neumann H (2009) Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J Off Publ Fed Am Soc Exp Biol 23:2502–2513. https://doi.org/10.1096/fj.08-123877
Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DG, Levy ML, Ransohoff RM, Gage FH, Glass CK (2017) An environment-dependent transcriptional network specifies human microglia identity. Science. https://doi.org/10.1126/science.aal3222
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19:504–516. https://doi.org/10.1038/nn.4222
Grundke-Iqbal I, Fleming J, Tung YC, Lassmann H, Iqbal K, Joshi JG (1990) Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol 81:105–110
Guerrero-Munoz MJ, Gerson J, Castillo-Carranza DL (2015) Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front Cell Neurosci 9:464. https://doi.org/10.3389/fncel.2015.00464
Guo JL, Lee VM-Y (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286:15317–15331. https://doi.org/10.1074/jbc.M110.209296
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1707-9
Gyorffy BA, Kun J, Torok G, Bulyaki E, Borhegyi Z, Gulyassy P, Kis V, Szocsics P, Micsonai A, Matko J, Drahos L, Juhasz G, Kekesi KA, Kardos J (2018) Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1722613115
Hansen DV, Hanson JE, Sheng M (2018) Microglia in Alzheimer’s disease. J Cell Biol 217:459–472. https://doi.org/10.1083/jcb.201709069
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan W-B, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9:1512–1519. https://doi.org/10.1038/nn1805
Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M (2014) Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13:60–69. https://doi.org/10.1111/acel.12149
Heidbreder A, Philipp K (2018) Anti-IgLON 5 Disease. Curr Treat Options Neurol 20:29. https://doi.org/10.1007/s11940-018-0515-4
Henkel K, Karitzky J, Schmid M, Mader I, Glatting G, Unger JW, Neumaier B, Ludolph AC, Reske SN, Landwehrmeyer GB (2004) Imaging of activated microglia with PET and [11C] PK 11195 in corticobasal degeneration. Mov Disord 19:817–821. https://doi.org/10.1002/mds.20040
Hickman S, Izzy S, Sen P, Morsett L, El Khoury J (2018) Microglia in neurodegeneration. Nat Neurosci 21:1359–1369. https://doi.org/10.1038/s41593-018-0242-x
Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. https://doi.org/10.1038/nn.3554
Hoglinger GU, Respondek G, Kovacs GG (2018) New classification of tauopathies. Rev Neurol (Paris). https://doi.org/10.1016/j.neurol.2018.07.001
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110:E3138–E3147. https://doi.org/10.1073/pnas.1301440110
Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W, Hol EM, Boddeke EWGM, Eggen BJL (2015) Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun 3:31. https://doi.org/10.1186/s40478-015-0203-5
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. https://doi.org/10.1126/science.aad8373
Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, Hyman BT (2018) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 15:269. https://doi.org/10.1186/s12974-018-1309-z
Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E (2012) Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci 32:15106–15111. https://doi.org/10.1523/JNEUROSCI.1167-12.2012
Huang Y, Xu Z, **ong S, Sun F, Qin G, Hu G, Wang J, Zhao L, Liang Y-X, Wu T, Lu Z, Humayun MS, So K-F, Pan Y, Li N, Yuan T-F, Rao Y, Peng B (2018) Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. https://doi.org/10.1038/s41593-018-0090-8
Huin V, Deramecourt V, Caparros-Lefebvre D, Maurage C-A, Duyckaerts C, Kovari E, Pasquier F, Buee-Scherrer V, Labreuche J, Behal H, Buee L, Dhaenens C-M, Sablonniere B (2016) The MAPT gene is differentially methylated in the progressive supranuclear palsy brain. Mov Disord 31:1883–1890. https://doi.org/10.1002/mds.26820
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. https://doi.org/10.1038/31508
Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, Ishiguro K, Hasegawa M, Yen S-H, Chishti MA, Harigaya Y, Abe K, Okamoto K, St George-Hyslop P, Westaway D (2005) Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol 166:521–531. https://doi.org/10.1016/S0002-9440(10)62274-2
Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M (2014) Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34:16180–16193. https://doi.org/10.1523/JNEUROSCI.3020-14.2014
Iqbal K, Liu F, Gong C-X (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12:15–27. https://doi.org/10.1038/nrneurol.2015.225
Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, Frenkel D, El Khoury J (2016) Megf10 Is a Receptor for C1Q That Mediates Clearance of Apoptotic Cells by Astrocytes. J Neurosci 36:5185–5192. https://doi.org/10.1523/JNEUROSCI.3850-15.2016
Ishizawa K, Dickson DW (2001) Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol 60:647–657
Ising C, Gallardo G, Leyns CEG, Wong CH, Stewart F, Koscal LJ, Roh J, Robinson GO, Remolina Serrano J, Holtzman DM (2017) AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J Exp Med. https://doi.org/10.1084/jem.20162125
Itagaki S, Akiyama H, Saito H, McGeer PL (1994) Ultrastructural localization of complement membrane attack complex (MAC)-like immunoreactivity in brains of patients with Alzheimer’s disease. Brain Res 645:78–84
Jadhav S, Avila J, Scholl M, Kovacs GG, Kovari E, Skrabana R, Evans LD, Kontsekova E, Malawska B, de Silva R, Buee L, Zilka N (2019) A walk through tau therapeutic strategies. Acta Neuropathol Commun 7:22. https://doi.org/10.1186/s40478-019-0664-z
Jadhav S, Cubinkova V, Zimova I, Brezovakova V, Madari A, Cigankova V, Zilka N (2015) Tau-mediated synaptic damage in Alzheimer’s disease. Transl Neurosci 6:214–226. https://doi.org/10.1515/tnsci-2015-0023
Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, Blennow K, Hansson O (2018) CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 91:e867–e877. https://doi.org/10.1212/WNL.0000000000006082
Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, LaFerla FM, Callahan LM, Federoff HJ, Bowers WJ (2008) Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol 173:1768–1782. https://doi.org/10.2353/ajpath.2008.080528
Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, Duimel H, Verheyen F, Kugler S, Van Leuven F (2011) Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol 179:2001–2015. https://doi.org/10.1016/j.ajpath.2011.06.025
Ji M, **e X-X, Liu D-Q, Yu X-L, Zhang Y, Zhang L-X, Wang S-W, Huang Y-R, Liu R-T (2018) Hepatitis B core VLP-based mis-disordered tau vaccine elicits strong immune response and alleviates cognitive deficits and neuropathology progression in Tau.P301S mouse model of Alzheimer’s disease and frontotemporal dementia. Alzheimers Res Ther 10:55. https://doi.org/10.1186/s13195-018-0378-7
Jiang T, Tan L, Zhu X-C, Zhou J-S, Cao L, Tan M-S, Wang H-F, Chen Q, Zhang Y-D, Yu J-T (2015) Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol Aging 36:3176–3186. https://doi.org/10.1016/j.neurobiolaging.2015.08.019
Jiang T, Zhang Y-D, Chen Q, Gao Q, Zhu X-C, Zhou J-S, Shi J-Q, Lu H, Tan L, Yu J-T (2016) TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105:196–206. https://doi.org/10.1016/j.neuropharm.2016.01.028
Jiang T, Zhang Y-D, Gao Q, Ou Z, Gong P-Y, Shi J-Q, Wu L, Zhou J-S (2018) TREM2 Ameliorates Neuronal Tau Pathology Through Suppression of Microglial Inflammatory Response. Inflammation. https://doi.org/10.1007/s10753-018-0735-5
Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, Kocher J-P, Petrucelli L, Fryer JD (2018) Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med 215:2235–2245. https://doi.org/10.1084/jem.20180653
Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Müller H-M, Nachman E, Steringer JP, Ruiz de Almodovar C, Nickel W, Jahn TR (2018) Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep 23:2039–2055. https://doi.org/10.1016/j.celrep.2018.04.056
Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI (2018) Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. https://doi.org/10.1007/s00401-018-1855-6
Kaufman SK, Thomas TL, Del Tredici K, Braak H, Diamond MI (2017) Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue. Acta Neuropathol Commun 5:41. https://doi.org/10.1186/s40478-017-0442-8
Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E (2018) Near-atomic model of microtubule-tau interactions. Science 360(6394):1242–1246
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169:1276–1290.e17. https://doi.org/10.1016/j.cell.2017.05.018
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287:19440–19451. https://doi.org/10.1074/jbc.M112.346072
Kim H, Park S, Cho H et al (2018) Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18f-av1451 positron emission tomography imaging. JAMA Neurol
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol 187:6539–6549. https://doi.org/10.4049/jimmunol.1100620
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci 25:8843–8853. https://doi.org/10.1523/JNEUROSCI.2868-05.2005
Klein RL, Dayton RD, Diaczynsky CG, Wang DB (2010) Pronounced microgliosis and neurodegeneration in aged rats after tau gene transfer. Neurobiol Aging 31:2091–2102. https://doi.org/10.1016/j.neurobiolaging.2008.12.002
Kobayashi E, Nakano M, Kubota K, Himuro N, Mizoguchi S, Chikenji T, Otani M, Mizue Y, Nagaishi K, Fujimiya M (2018) Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci Rep 8:1712. https://doi.org/10.1038/s41598-018-19442-7
Koriath C, Lashley T, Taylor W, Druyeh R, Dimitriadis A, Denning N, Williams J, Warren JD, Fox NC, Schott JM, Rowe JB, Collinge J, Rohrer JD, Mead S (2019) ApoE4 lowers age at onset in patients with frontotemporal dementia and tauopathy independent of amyloid-β copathology. Alzheimer’s Dement (Amsterdam, Netherlands) 11:277–280. https://doi.org/10.1016/j.dadm.2019.01.010
Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, Novak M (2011) Misfolded truncated protein tau induces innate immune response via MAPK pathway. J Immunol 187:2732–2739. https://doi.org/10.4049/jimmunol.1100216
Kovac A, Zilkova M, Deli MA, Zilka N, Novak M (2009) Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis 18:897–906. https://doi.org/10.3233/JAD-2009-1197
Kovacech B, Novak M (2010) Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer’s disease. Curr Alzheimer Res 7:708–716. https://doi.org/10.2174/156720510793611556
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47:566–581.e9. https://doi.org/10.1016/j.immuni.2017.08.008
Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, Riether C, Meyer U, Knuesel I (2012) Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation 9:151. https://doi.org/10.1186/1742-2094-9-151
Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kugler S, Van Leuven F, Cuadrado A (2014) Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 137:78–91. https://doi.org/10.1093/brain/awt323
Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, Dong Y, Legeron F-P, Leboucher A, Burnouf S, Faivre E, Carvalho K, Caillierez R, Zommer N, Demeyer D, Jouy N, Sazdovitch V, Schraen-Maschke S, Delarasse C, Buee L, Blum D (2017) Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140:184–200. https://doi.org/10.1093/brain/aww270
Le Guennec K, Quenez O, Nicolas G, Wallon D, Rousseau S, Richard A-C, Alexander J, Paschou P, Charbonnier C, Bellenguez C, Grenier-Boley B, Lechner D, Bihoreau M-T, Olaso R, Boland A, Meyer V, Deleuze J-F, Amouyel P, Munter HM, Bourque G, Lathrop M, Frebourg T, Redon R, Letenneur L, Dartigues J-F, Martinaud O, Kalev O, Mehrabian S, Traykov L, Strobel T, Le Ber I, Caroppo P, Epelbaum S, Jonveaux T, Pasquier F, Rollin-Sillaire A, Genin E, Guyant-Marechal L, Kovacs GG, Lambert J-C, Hannequin D, Campion D, Rovelet-Lecrux A (2017) 17q21.31 duplication causes prominent tau-related dementia with increased MAPT expression. Mol Psychiatry 22:1119–1125. https://doi.org/10.1038/mp.2016.226
Lee DC, Rizer J, Selenica M-LB, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D (2010) LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation 7:56. https://doi.org/10.1186/1742-2094-7-56
Lee S, Xu G, Jay TR, Bhatta S, Kim K-W, Jung S, Landreth GE, Ransohoff RM, Lamb BT (2014) Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J Neurosci 34:12538–12546. https://doi.org/10.1523/JNEUROSCI.0853-14.2014
Lee S-H, Le Pichon CE, Adolfsson O, Gafner V, Pihlgren M, Lin H, Solanoy H, Brendza R, Ngu H, Foreman O, Chan R, Ernst JA, DiCara D, Hotzel I, Srinivasan K, Hansen DV, Atwal J, Lu Y, Bumbaca D, Pfeifer A, Watts RJ, Muhs A, Scearce-Levie K, Ayalon G (2016) Antibody-Mediated Targeting of Tau In Vivo Does Not Require Effector Function and Microglial Engagement. Cell Rep. https://doi.org/10.1016/j.celrep.2016.06.099
Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, Stevens B (2018) CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 100:120–134.e6. https://doi.org/10.1016/j.neuron.2018.09.017
Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF-alpha Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 90:483–491. https://doi.org/10.1016/j.neuron.2016.03.030
Leyns CEG, Holtzman DM (2017) Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener 12:50. https://doi.org/10.1186/s13024-017-0192-x
Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM (2017) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1710311114
Li J-G, Chu J, Pratico D (2018) Downregulation of autophagy by 12/15Lipoxygenase worsens the phenotype of an Alzheimer’s disease mouse model with plaques, tangles, and memory impairments. Mol Psychiatry. https://doi.org/10.1038/s41380-018-0268-1
Li Y, Liu L, Barger SW, Griffin WST (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23:1605–1611
Lian H, Litvinchuk A, Chiang AC-A, Aithmitti N, Jankowsky JL, Zheng H (2016) Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci 36:577–589. https://doi.org/10.1523/JNEUROSCI.2117-15.2016
Lian H, Yang L, Cole A, Sun L, Chiang AC-A, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu H-C, Zheng H (2015) NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85:101–115. https://doi.org/10.1016/j.neuron.2014.11.018
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung W-S, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Linnartz B, Kopatz J, Tenner AJ, Neumann H (2012) Sialic Acid on the Neuronal Glycocalyx Prevents Complement C1 Binding and Complement Receptor-3-Mediated Removal by Microglia. J Neurosci 32:946–952. https://doi.org/10.1523/JNEUROSCI.3830-11.2012
Litvinchuk A, Wan Y-W, Swartzlander DB, Chen F, Cole A, Propson NE, Wang Q, Zhang B, Liu Z, Zheng H (2018) Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron. https://doi.org/10.1016/j.neuron.2018.10.031
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang H-Y, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam M-M, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RVJ, Paz JT, Barres BA, Huang EJ (2016) Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165:921–935. https://doi.org/10.1016/j.cell.2016.04.001
Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM (2015) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5:11161. https://doi.org/10.1038/srep11161
Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, Bryan RM, Attwell D (2018) Microglial Ramification, Surveillance, and Interleukin-1beta Release Are Regulated by the Two-Pore Domain K(+) Channel THIK-1. Neuron 97:299–312.e6. https://doi.org/10.1016/j.neuron.2017.12.002
Majerova P, Garruto RM, Kovac A (2018) Cerebrovascular inflammation is associated with tau pathology in Guam parkinsonism dementia. J Neural Transm. https://doi.org/10.1007/s00702-018-1883-3
Majerova P, Michalicova A, Cente M, Hanes J, Vegh J, Kittel A, Kosikova N, Cigankova V, Mihaljevic S, Jadhav S, Kovac A (2019) Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS One 14:e0217216. https://doi.org/10.1371/journal.pone.0217216
Majerova P, Zilkova M, Kazmerova Z, Kovac A, Paholikova K, Kovacech B, Zilka N, Novak M (2014) Microglia display modest phagocytic capacity for extracellular tau oligomers. J Neuroinflammation 11:161. https://doi.org/10.1186/s12974-014-0161-z
Maphis N, Jiang S, Xu G, Kokiko-Cochran ON, Roy SM, Van Eldik LJ, Watterson DM, Lamb BT, Bhaskar K (2016) Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res Ther 8:54. https://doi.org/10.1186/s13195-016-0221-y
Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, Bhaskar K (2015) Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci 9:196. https://doi.org/10.3389/fnins.2015.00196
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138:1738–1755. https://doi.org/10.1093/brain/awv081
Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan Y-W, Bajaj L, Bader DA, Lee VMY, Trojanowski JQ, Liu Z, Sardiello M, Zheng H (2018) TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med 215:2355–2377. https://doi.org/10.1084/jem.20172158
Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, Ozkaya N, Pastore A, Schwabenland M, Chung YR, Rosenblum MK, Prinz M, Abdel-Wahab O, Geissmann F (2017) A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature. https://doi.org/10.1038/nature23672
Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS, Perona M, Desai R, Smith KJ, Latcham J, Fulleylove M, Richardson JC, Hardy J, Edwards FA (2015) A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10:633–644. https://doi.org/10.1016/j.celrep.2014.12.041
Mattsson N, Eriksson O, Lindberg O, Scholl M, Lampinen B, Nilsson M, Insel PS, Lautner R, Strandberg O, van Westen D, Zetterberg H, Blennow K, Palmqvist S, Stomrud E, Hansson O (2018) Effects of APOE epsilon4 on neuroimaging, cerebrospinal fluid biomarkers, and cognition in prodromal Alzheimer’s disease. Neurobiol Aging 71:81–90. https://doi.org/10.1016/j.neurobiolaging.2018.07.003
Mattsson N, Ossenkoppele R, Smith R, Strandberg O, Ohlsson T, Jogi J, Palmqvist S, Stomrud E, Hansson O (2018) Greater tau load and reduced cortical thickness in APOE epsilon4-negative Alzheimer’s disease: a cohort study. Alzheimers Res Ther 10:77. https://doi.org/10.1186/s13195-018-0403-x
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Immune system response in Alzheimer’s disease. Can J Neurol Sci 16:516–527
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346
McQuade A, Blurton-Jones M (2019) Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J Mol Biol 431:1805–1817. https://doi.org/10.1016/j.jmb.2019.01.045
Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, Uronen R-L, Huttunen HJ (2018) Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep 25:2027–2035.e4. https://doi.org/10.1016/j.celrep.2018.10.078
Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, Seeley WW, Mucke L, Nelson RB, Akassoglou K (2019) Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 101:1099–1108.e6. https://doi.org/10.1016/j.neuron.2019.01.014
Merlini M, Wanner D, Nitsch RM (2016) Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131:737–752. https://doi.org/10.1007/s00401-016-1560-2
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, Cormier KA, Kubilus CA, Martin BM, Murphy L, Baugh CM, Montenigro PH, Chaisson CE, Tripodis Y, Kowall NW, Weuve J, McClean MD, Cantu RC, Goldstein LE, Katz DI, Stern RA, Stein TD, McKee AC (2017) Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 318:360–370. https://doi.org/10.1001/jama.2017.8334
Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, Joachimiak LA, Diamond MI (2018) Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 7:e36584. https://doi.org/10.7554/eLife.36584
Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, Koizumi S, Moorhouse AJ, Yoshimura Y, Nabekura J (2016) Microglia contact induces synapse formation in develo** somatosensory cortex. Nat Commun 7:12540. https://doi.org/10.1038/ncomms12540
Montgomery SL, Mastrangelo MA, Habib D, Narrow WC, Knowlden SA, Wright TW, Bowers WJ (2011) Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: implications for chronic pan-TNF-alpha suppressive therapeutic strategies in the brain. Am J Pathol 179:2053–2070. https://doi.org/10.1016/j.ajpath.2011.07.001
Montgomery SL, Narrow WC, Mastrangelo MA, Olschowka JA, O’Banion MK, Bowers WJ (2013) Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic Alzheimer disease mice leads to significant modulation of amyloid- and Tau-related pathologies. Am J Pathol 182:2285–2297. https://doi.org/10.1016/j.ajpath.2013.02.030
Morales I, Jimenez JM, Mancilla M, Maccioni RB (2013) Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis 37:849–856. https://doi.org/10.3233/JAD-131843
Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, Nabekura J, Sato K, Okajima F, Takebayashi H, Okano H, Koizumi S (2017) Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun 8:28. https://doi.org/10.1038/s41467-017-00037-1
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM, Mandelkow E-M, Mandelkow E, Buee L, Goedert M, Brion J-P (2017) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5:99. https://doi.org/10.1186/s40478-017-0488-7
Muth C, Hartmann A, Sepulveda-Falla D, Glatzel M, Krasemann S (2019) Phagocytosis of Apoptotic Cells Is Specifically Upregulated in ApoE4 Expressing Microglia in vitro. Front Cell Neurosci 13:181. https://doi.org/10.3389/fncel.2019.00181
Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY (2017) Pathological tau strains from human brains recapitulate the diversity of tauopathies in non-transgenic mouse brain. J Neurosci. https://doi.org/10.1523/JNEUROSCI.1230-17.2017
Nash KR, Lee DC, Hunt JBJ, Morganti JM, Selenica M-L, Moran P, Reid P, Brownlow M, Guang-Yu Yang C, Savalia M, Gemma C, Bickford PC, Gordon MN, Morgan D (2013) Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging 34:1540–1548. https://doi.org/10.1016/j.neurobiolaging.2012.12.011
Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, Kayed R (2017) Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J Alzheimers Dis 55:1083–1099. https://doi.org/10.3233/JAD-160912
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 308:1314–1318
Nisbet RM, Van der Jeugd A, Leinenga G, Evans HT, Janowicz PW, Gotz J (2017) Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain. https://doi.org/10.1093/brain/awx052
Nistico R, Salter E, Nicolas C, Feligioni M, Mango D, Bortolotto ZA, Gressens P, Collingridge GL, Peineau S (2017) Synaptoimmunology - roles in health and disease. Mol Brain 10:26. https://doi.org/10.1186/s13041-017-0308-9
Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH (2009) Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol 23:739–750. https://doi.org/10.1096/fj.08-113795
Nobuhara CK, DeVos SL, Commins C, Wegmann S, Moore BD, Roe AD, Costantino I, Frosch MP, Pitstick R, Carlson GA, Hock C, Nitsch RM, Montrasio F, Grimm J, Cheung AE, Dunah AW, Wittmann M, Bussiere T, Weinreb PH, Hyman BT, Takeda S (2017) Tau Antibody-Targeting Pathological Species Block Neuronal Uptake and Interneuron Propagation of Tau in Vitro. Am J Pathol. https://doi.org/10.1016/j.ajpath.2017.01.022
Novak P, Kontsekova E, Zilka N, Novak M (2018) Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front Neurosci 12:798. https://doi.org/10.3389/fnins.2018.00798
Odawara T, Iseki E, Kosaka K, Akiyama H, Ikeda K, Yamamoto T (1995) Investigation of tau-2 positive microglia-like cells in the subcortical nuclei of human neurodegenerative disorders. Neurosci Lett 192:145–148
Ojala JO, Sutinen EM, Salminen A, Pirttila T (2008) Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J Neuroimmunol 205:86–93. https://doi.org/10.1016/j.jneuroim.2008.09.012
Olah M, Patrick E, Villani A-C, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM (2018) A transcriptomic atlas of aged human microglia. Nat Commun 9:539. https://doi.org/10.1038/s41467-018-02926-5
Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, O’Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139:1551–1567. https://doi.org/10.1093/brain/aww027
Overmyer M, Helisalmi S, Soininen H, Laakso M, Riekkinen PS, Alafuzoff I (1999) Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol 97:383–392
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. https://doi.org/10.1126/science.1202529
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan W-B (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. https://doi.org/10.1016/j.cell.2013.11.030
Patel TK, Habimana-Griffin L, Gao X, Xu B, Achilefu S, Alitalo K, McKee CA, Sheehan PW, Musiek ES, **ong C, Coble D, Holtzman DM (2019) Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol Neurodegener 14:11. https://doi.org/10.1186/s13024-019-0312-x
Paulus W, Bancher C, Jellinger K (1993) Microglial reaction in Pick’s disease. Neurosci Lett 161:89–92
Perea JR, Avila J, Bolos M (2018) Dephosphorylated rather than hyperphosphorylated Tau triggers a pro-inflammatory profile in microglia through the p38 MAPK pathway. Exp Neurol 310:14–21. https://doi.org/10.1016/j.expneurol.2018.08.007
Piacentini R, Li Puma DD, Mainardi M, Lazzarino G, Tavazzi B, Arancio O, Grassi C (2017) Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 65:1302–1316. https://doi.org/10.1002/glia.23163
Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, Shuken SR, Gate D, Scott M, Khatri P, Luo J, Bertozzi CR, Bassik MC, Wyss-Coray T (2019) CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568:187–192. https://doi.org/10.1038/s41586-019-1088-4
Polanco JC, Scicluna BJ, Hill AF, Gotz J (2016) Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J Biol Chem 291:12445–12466. https://doi.org/10.1074/jbc.M115.709485
Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP (2013) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14:389–394. https://doi.org/10.1038/embor.2013.15
Prada I, Gabrielli M, Turola E, Iorio A, D’Arrigo G, Parolisi R, De Luca M, Pacifici M, Bastoni M, Lombardi M, Legname G, Cojoc D, Buffo A, Furlan R, Peruzzi F, Verderio C (2018) Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1803-x
Probst A, Ulrich J, Heitz PU (1982) Senile dementia of Alzheimer type: astroglial reaction to extracellular neurofibrillary tangles in the hippocampus. An immunocytochemical and electron-microscopic study. Acta Neuropathol 57:75–79
Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB (2004) Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res 295:245–257. https://doi.org/10.1016/j.yexcr.2004.01.002
Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM (2017) Prophylactic Active Tau Immunization Leads to Sustained Reduction in Both Tau and Amyloid-beta Pathologies in 3xTg Mice. Sci Rep 7:17034. https://doi.org/10.1038/s41598-017-17313-1
Rasmussen MK, Mestre H, Nedergaard M (2018) The glymphatic pathway in neurological disorders. Lancet Neurol 17:1016–1024. https://doi.org/10.1016/S1474-4422(18)30318-1
Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, Hsieh-Wilson LC, Kampmann M, Kosik KS (2018) Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep 8:6382. https://doi.org/10.1038/s41598-018-24904-z
Rodriguez-Callejas JD, Fuchs E, Perez-Cruz C (2016) Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front Aging Neurosci 8:315. https://doi.org/10.3389/fnagi.2016.00315
Roe AD, Staup MA, Serrats J, Sawchenko PE, Rissman RA (2011) Lipopolysaccharide-induced tau phosphorylation and kinase activity--modulation, but not mediation, by corticotropin-releasing factor receptors. Eur J Neurosci 34:448–456. https://doi.org/10.1111/j.1460-9568.2011.07764.x
Romero-Molina C, Navarro V, Sanchez-Varo R, Jimenez S, Fernandez-Valenzuela JJ, Sanchez-Mico MV, Munoz-Castro C, Gutierrez A, Vitorica J, Vizuete M (2018) Distinct Microglial Responses in Two Transgenic Murine Models of TAU Pathology. Front Cell Neurosci 12:421. https://doi.org/10.3389/fncel.2018.00421
Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O (2006) Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol 63:1459–1467. https://doi.org/10.1001/archneur.63.10.1459
Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao C-C, Ardura-Fabregat A, de Lima KA, Gutierrez-Vazquez C, Hewson P, Staszewski O, Blain M, Healy L, Neziraj T, Borio M, Wheeler M, Dragin LL, Laplaud DA, Antel J, Alvarez JI, Prinz M, Quintana FJ (2018) Microglial control of astrocytes in response to microbial metabolites. Nature 557:724–728. https://doi.org/10.1038/s41586-018-0119-x
Rozenstein-Tsalkovich L, Grigoriadis N, Lourbopoulos A, Nousiopoulou E, Kassis I, Abramsky O, Karussis D, Rosenmann H (2013) Repeated immunization of mice with phosphorylated-tau peptides causes neuroinflammation. Exp Neurol 248:451–456. https://doi.org/10.1016/j.expneurol.2013.07.006
Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, Edinger AL, Jung S, Rossner MJ, Simons M (2016) Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci 19:995–998. https://doi.org/10.1038/nn.4325
Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NCY, Hall GF (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287:3842–3849. https://doi.org/10.1074/jbc.M111.277061
Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, Trujillo-Estrada L, Davila JC, Vizuete M, Gutierrez A, Vitorica J (2016) Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol 132:897–916. https://doi.org/10.1007/s00401-016-1630-5
Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, Hoque N, DeCarr L, Keenan S, Lin A, Cao Y, Snyder B, Zhang B, Nitla M, Hirschfeld G, Barrezueta N, Polson C, Wes P, Rangan VS, Cacace A, Albright CF, Meredith J, Trojanowski JQ, Lee VM-Y, Brunden KR, Ahlijanian M (2015) Passive Immunization with Phospho-Tau Antibodies Reduces Tau Pathology and Functional Deficits in Two Distinct Mouse Tauopathy Models. PLoS One 10:e0125614. https://doi.org/10.1371/journal.pone.0125614
Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM (2012) Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem 287:20522–20533. https://doi.org/10.1074/jbc.M111.323279
Sasaki A, Kawarabayashi T, Murakami T, Matsubara E, Ikeda M, Hagiwara H, Westaway D, George-Hyslop PS, Shoji M, Nakazato Y (2008) Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res 1214:159–168. https://doi.org/10.1016/j.brainres.2008.02.084
Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, Zhan L, Cooper YA, Davalos D, Akassoglou K, Coppola G, Gan L (2018) Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc Natl Acad Sci
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Scheff SW, Price DA, Ansari MA, Roberts KN, Schmitt FA, Ikonomovic MD, Mufson EJ (2015) Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis 43:1073–1090. https://doi.org/10.3233/JAD-141518
Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ (2011) Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 24:547–557. https://doi.org/10.3233/JAD-2011-101782
Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2017) Alzheimer’s disease. Lancet 388:505–517. https://doi.org/10.1016/S0140-6736(15)01124-1
Schmid AW, Lynch MA, Herron CE (2009) The effects of IL-1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus 19:670–676. https://doi.org/10.1002/hipo.20542
Schwab C, Steele JC, McGeer PL (1996) Neurofibrillary tangles of Guam parkinson-dementia are associated with reactive microglia and complement proteins. Brain Res 707:196–205
Selenica M-LB, Davtyan H, Housley SB, Blair LJ, Gillies A, Nordhues BA, Zhang B, Liu J, Gestwicki JE, Lee DC, Gordon MN, Morgan D, Dickey CA (2014) Epitope analysis following active immunization with tau proteins reveals immunogens implicated in tau pathogenesis. J Neuroinflammation 11:152. https://doi.org/10.1186/s12974-014-0152-0
Serrano-Pozo A, Mielke ML, Gomez-Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol 179:1373–1384. https://doi.org/10.1016/j.ajpath.2011.05.047
Sharma AM, Thomas TL, Woodard DR, Kashmer OM, Diamond MI (2018) Tau monomer encodes strains. Elife 7. https://doi.org/10.7554/eLife.37813
Sheffield LG, Marquis JG, Berman NE (2000) Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett 285:165–168
Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, Goux WJ, Lee V, Johnson GV, Webster SD, Cooper NR, Bradt B, Rogers J (2001) Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett 305:165–168
Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS (2001) Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int 39:341–348
Sheng JG, Mrak RE, Griffin WS (1997) Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol 56:285–290
Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart J-C, Caldarone BJ, Stevens B, Lemere CA (2015) Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci 35:13029–13042. https://doi.org/10.1523/JNEUROSCI.1698-15.2015
Shi Y, Holtzman DM (2018) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. https://doi.org/10.1038/s41577-018-0051-1
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, **ong C, Del-Aguila JL, Morris JC, Cruchaga C, Initiative ADN, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549(7673):523–527
Sidoryk-Wegrzynowicz M, Gerber YN, Ries M, Sastre M, Tolkovsky AM, Spillantini MG (2017) Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol Commun 5:89. https://doi.org/10.1186/s40478-017-0478-9
Singhrao SK, Neal JW, Gasque P, Morgan BP, Newman GR (1996) Role of complement in the aetiology of Pick’s disease? J Neuropathol Exp Neurol 55:578–593
Sipe GO, Lowery RL, Tremblay M-E, Kelly EA, Lamantia CE, Majewska AK (2016) Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun 7:10905. https://doi.org/10.1038/ncomms10905
Socodato R, Portugal CC, Canedo T, Henriques J, Vaz SH, Magalhaes JD, Silva CM, Baptista FI, Alves RL, Nogueira J, Coelho-Santos V, Silva AP, Paes-de-Carvalho R, Magalhaes A, Summavielle T, Brakebusch C, Sebastiao AM, Ambrosio AF, Relvas JB (2017) Genetic ablation of RhoA in adult microglia causes synapse and neuronal loss. bioRxiv
Sotiropoulos I, Galas M-C, Silva JM, Skoulakis E, Wegmann S, Maina MB, Blum D, Sayas CL, Mandelkow E-M, Mandelkow E, Spillantini MG, Sousa N, Avila J, Medina M, Mudher A, Buee L (2017) Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun 5:91. https://doi.org/10.1186/s40478-017-0489-6
Spencer B, Brüschweiler S, Sealey-Cardona M, Rockenstein E, Adame A, Florio J, Mante M, Trinh I, Rissman RA, Konrat R, Masliah E (2018) Selective targeting of 3 repeat Tau with brain penetrating single chain antibodies for the treatment of neurodegenerative disorders. Acta Neuropathol 136:69–87. https://doi.org/10.1007/s00401-018-1869-0
Stancu I-C, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brone B, Huaux F, Octave J-N, Terwel D, Dewachter I (2019) Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137:599–617. https://doi.org/10.1007/s00401-018-01957-y
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai H-H, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA (2013) A Dramatic Increase of C1q Protein in the CNS during Normal Aging. J Neurosci 33:13460–13474. https://doi.org/10.1523/JNEUROSCI.1333-13.2013
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA (2000) Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am J Pathol 156:489–499. https://doi.org/10.1016/S0002-9440(10)64753-0
Stopschinski BE, Holmes BB, Miller GM, Manon VA, Vaquer-Alicea J, Prueitt WL, Hsieh-Wilson LC, Diamond MI (2018) Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau vs. alpha-synuclein and beta-amyloid aggregates. J Biol Chem. https://doi.org/10.1074/jbc.RA117.000378
Stozicka Z, Zilka N, Novak P, Kovacech B, Bugos O, Novak M (2010) Genetic background modifies neurodegeneration and neuroinflammation driven by misfolded human tau protein in rat model of tauopathy: implication for immunomodulatory approach to Alzheimer’s disease. J Neuroinflammation 7:64. https://doi.org/10.1186/1742-2094-7-64
Streit WJ, Braak H, Del Tredici K, Leyh J, Lier J, Khoshbouei H, Eisenloffel C, Muller W, Bechmann I (2018) Microglial activation occurs late during preclinical Alzheimer’s disease. Glia. https://doi.org/10.1002/glia.23510
Streit WJ, Braak H, Xue Q-S, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118:475–485. https://doi.org/10.1007/s00401-009-0556-6
Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212. https://doi.org/10.1002/glia.10319
Svahn AJ, Don EK, Badrock AP, Cole NJ, Graeber MB, Yerbury JJ, Chung R, Morsch M (2018) Nucleo-cytoplasmic transport of TDP-43 studied in real time: impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons. Acta Neuropathol 136:445–459. https://doi.org/10.1007/s00401-018-1875-2
Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM (2011) Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol 178:2811–2822. https://doi.org/10.1016/j.ajpath.2011.02.012
Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R, Buee L, Colin M (2016) Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 4:117. https://doi.org/10.1186/s40478-016-0386-4
Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, Hickman DT, Adolfsson O, Chuard N, Ndao DM, Borghgraef P, Devijver H, Van Leuven F, Pfeifer A, Muhs A (2013) Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau. P301L mice that model tauopathy. PLoS One 8:e72301. https://doi.org/10.1371/journal.pone.0072301
Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, Bechmann I (2016) Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia 64:1562–1572. https://doi.org/10.1002/glia.23024
Tremblay M-E, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8:e1000527. https://doi.org/10.1371/journal.pbio.1000527
Tseng J-H, **e L, Song S, **e Y, Allen L, Ajit D, Hong J-S, Chen X, Meeker RB, Cohen TJ (2017) The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep 20:2169–2183. https://doi.org/10.1016/j.celrep.2017.07.082
Vagnozzi AN, Giannopoulos PF, Pratico D (2017) Brain 5-lipoxygenase over-expression worsens memory, synaptic integrity, and tau pathology in the P301S mice. Aging Cell. https://doi.org/10.1111/acel.12695
Vagnozzi AN, Giannopoulos PF, Pratico D (2017) The direct role of 5-lipoxygenase on tau pathology, synaptic integrity and cognition in a mouse model of tauopathy. Transl Psychiatry 7:1288. https://doi.org/10.1038/s41398-017-0017-2
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, Molofsky AV (2018) Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359:1269–1273. https://doi.org/10.1126/science.aal3589
Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, Kennedy M, Kozauer N, Margolin RA, Molinuevo JL, Mueller R, Ransohoff RM, Wilcock DM, Bain L, Bales K (2016) The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv 2:99–109. https://doi.org/10.1016/j.trci.2016.05.001
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, Vandamme N, De Schepper S, Van Isterdael G, Scott CL, Aerts J, Berx G, Boeckxstaens GE, Vandenbroucke RE, Vereecke L, Moechars D, Guilliams M, Van Ginderachter JA, Saeys Y, Movahedi K (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22:1021–1035. https://doi.org/10.1038/s41593-019-0393-4
Veerhuis R, Nielsen HM, Tenner AJ (2011) Complement in the brain. Mol Immunol 48:1592–1603. https://doi.org/10.1016/j.molimm.2011.04.003
Vitale F, Giliberto L, Ruiz S, Steslow K, Marambaud P, d’Abramo C (2018) Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun 6:82. https://doi.org/10.1186/s40478-018-0585-2
von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem 276:48165–48174. https://doi.org/10.1074/jbc.M105196200
von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306) VQIVYK (311)) forming beta structure. Proc Natl Acad Sci U S A 97:5129–5134
Wang H, Li Y, Ryder JW, Hole JT, Ebert PJ, Airey DC, Qian H-R, Logsdon B, Fisher A, Ahmed Z, Murray TK, Cavallini A, Bose S, Eastwood BJ, Collier DA, Dage JL, Miller BB, Merchant KM, O’Neill MJ, Demattos RB (2018) Genome-wide RNAseq study of the molecular mechanisms underlying microglia activation in response to pathological tau perturbation in the rTg4510 tau transgenic animal model. Mol Neurodegener 13:65. https://doi.org/10.1186/s13024-018-0296-y
Wang L, Jiang Q, Chu J, Lin L, Li X-G, Chai G-S, Wang Q, Wang J-Z, Tian Q (2013) Expression of Tau40 induces activation of cultured rat microglial cells. PLoS One 8:e76057. https://doi.org/10.1371/journal.pone.0076057
Wang Q, Wu J, Rowan MJ, Anwyl R (2005) Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci 22:2827–2832. https://doi.org/10.1111/j.1460-9568.2005.04457.x
Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, Wang M, Qian HH, Badea TC, Diamond JS, Gan W-B, Roger JE, Wong WT (2016) Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. J Neurosci 36:2827–2842. https://doi.org/10.1523/JNEUROSCI.3575-15.2016
Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, Mandelkow E-M (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12:5. https://doi.org/10.1186/s13024-016-0143-y
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21. https://doi.org/10.1038/nrn.2015.1
Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J (1997) Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging 18:415–421
Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, Schwab Y, Gross CT (2018) Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9:1228. https://doi.org/10.1038/s41467-018-03566-5
Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE (2013) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288:1856–1870. https://doi.org/10.1074/jbc.M112.394528
Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RACM, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. https://doi.org/10.1038/nn.4328
Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2015) Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2:278–288. https://doi.org/10.1002/acn3.176
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. https://doi.org/10.1016/j.neuron.2013.07.046
Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, Bontha SV, Srikakulapu P, Beer M, Megens RTA, Steffens S, Hildner M, Halder LD, Eckstein H-H, Pelisek J, Herms J, Roeber S, Arzberger T, Borodovsky A, Habenicht L, Binder CJ, Weber C, Zipfel PF, Skerka C, Habenicht AJR (2019) ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med 25:496–506. https://doi.org/10.1038/s41591-018-0336-8
Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM-Y (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. https://doi.org/10.1016/j.neuron.2007.01.010
Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, Masone A, Corbelli A, Smith DH, Menon DK, Stocchetti N, Fiordaliso F, De Simoni M-G, Stewart W, Chiesa R (2018) Induction of a transmissible tau pathology by traumatic brain injury. Brain 141:2685–2699. https://doi.org/10.1093/brain/awy193
Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, Price JL (2005) Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord 19:55–66
Zempel H, Mandelkow E (2014) Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci 37:721–732. https://doi.org/10.1016/j.tins.2014.08.004
Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17:400–406. https://doi.org/10.1038/nn.3641
Zhao N, Liu C-C, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, Attrebi ON, Petrucelli L, Fryer JD, Wszolek ZK, Graff-Radford NR, Caselli RJ, Sanchez-Contreras MY, Rademakers R, Murray ME, Koga S, Dickson DW, Ross OA, Bu G (2018) APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun 9:4388. https://doi.org/10.1038/s41467-018-06783-0
Zhong Z, Grasso L, Sibilla C, Stevens TJ, Barry N, Bertolotti A (2018) Prion-like protein aggregates exploit the RHO GTPase to cofilin-1 signaling pathway to enter cells. EMBO J 37. https://doi.org/10.15252/embj.201797822
Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, Rolkova G, Kontsekova E, Novak M (2006) Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 580:3582–3588. https://doi.org/10.1016/j.febslet.2006.05.029
Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M (2012) Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation 9:47. https://doi.org/10.1186/1742-2094-9-47
Zilka N, Stozicka Z, Cente M, Kazmerova Z, Kovacech B, Novak M (2012) Immunomodulation of memory-impairing protein tau in Alzheimer’s disease. Neurodegener Dis 10:242–245. https://doi.org/10.1159/000333125
Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M (2009) Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol 209:16–25. https://doi.org/10.1016/j.jneuroim.2009.01.013
Acknowledgements
The authors would like to thank Norbert Zilka for his feedback on the manuscript.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 676144 (Synaptic Dysfunction in Alzheimer Disease, SyDAD).
Author information
Authors and Affiliations
Contributions
TV conceived the topic and drafted the manuscript. TV, AM & TH made figures. TH critically read and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
TV and TH are employees of Axon Neuroscience R&D Services SE.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Vogels, T., Murgoci, AN. & Hromádka, T. Intersection of pathological tau and microglia at the synapse. acta neuropathol commun 7, 109 (2019). https://doi.org/10.1186/s40478-019-0754-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-019-0754-y