
Abstract
Background
Plants live with diverse microbial communities which profoundly affect multiple facets of host performance, but if and how host development impacts the assembly, functions and microbial interactions of crop microbiomes are poorly understood. Here we examined both bacterial and fungal communities across soils, epiphytic and endophytic niches of leaf and root, and plastic leaf of fake plant (representing environment-originating microbes) at three developmental stages of maize at two contrasting sites, and further explored the potential function of phylloplane microbiomes based on metagenomics.
Results
Our results suggested that plant developmental stage had a much stronger influence on the microbial diversity, composition and interkingdom networks in plant compartments than in soils, with the strongest effect in the phylloplane. Phylloplane microbiomes were co-shaped by both plant growth and seasonal environmental factors, with the air (represented by fake plants) as its important source. Further, we found that bacterial communities in plant compartments were more strongly driven by deterministic processes at the early stage but a similar pattern was for fungal communities at the late stage. Moreover, bacterial taxa played a more important role in microbial interkingdom network and crop yield prediction at the early stage, while fungal taxa did so at the late stage. Metagenomic analyses further indicated that phylloplane microbiomes possessed higher functional diversity at the early stage than the late stage, with functional genes related to nutrient provision enriched at the early stage and N assimilation and C degradation enriched at the late stage. Coincidently, more abundant beneficial bacterial taxa like Actinobacteria, Burkholderiaceae and Rhizobiaceae in plant microbiomes were observed at the early stage, but more saprophytic fungi at the late stage.
Conclusions
Our results suggest that host developmental stage profoundly influences plant microbiome assembly and functions, and the bacterial and fungal microbiomes take a differentiated ecological role at different stages of plant development. This study provides empirical evidence for host exerting strong effect on plant microbiomes by deterministic selection during plant growth and development. These findings have implications for the development of future tools to manipulate microbiome for sustainable increase in primary productivity.
Video Abstract
Similar content being viewed by others


Background
Plants live with large and diverse prokaryotes and eukaryotes (i.e. plant microbiomes) which have coevolved with their hosts and profoundly impact a range of aspects of plant performance [1,2,3,4]. For example, some beneficial bacteria and fungi like nitrogen fixer, antagonistic bacteria and mycorrhizal fungi in the rhizosphere and plant compartments can deeply influence plant growth and health via promoting nutrient acquisition, protecting against pathogen attacks, and increasing tolerance to environmental stress [5,6,7,8]. Recent studies suggested that plant microbiome assembly and host health are largely influenced by complex and dynamic interactions between the host, microbes, and the environment, but the ecological processes that govern plant-microbiome-environment interactions remain poorly understood [3, 9, 10]. A better understanding of the mechanisms and temporal dynamics of plant microbiome assembly, functions and co-occurrence networks is of significant importance for the development of microbiome-based solutions for sustainable crop production systems [11,12,13,14].
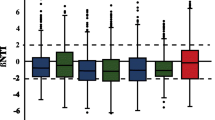
Assembly of plant microbiomes starts soon after sowing and develops with plant growth under the influence of deterministic (e.g. selection mediated by biotic and abiotic factors) and stochastic (e.g. random dispersal and drift events) processes [2, 7, 15]. In addition to microbial inheritance and vertical transmission from seed [2, 16, 17], microbes can colonize different plant compartments through dispersal from soil, air and nearby plants, and then form a dynamic community under the integrative effects of host and environmental factors [4, 7, 15, 18, 19]. On the one hand, the plant host has strong selection effects on its microbiomes via host immune system, genetic networks and plant exudates [20,21,22,23,24]. On the other hand, multiple environmental factors such as climate, edaphic properties (e.g. soil pH and nutrients) and human perturbations (e.g. agricultural management regimes) also play important roles in driving plant microbiome assembly [25,26,27,28,29]. It has been reported that plant microbiomes were mainly determined by compartment niche and host species at the plant level, with the phylloplane and rhizoplane acting as an important interface between the host and the environment [30,31,32, The field experiments were located in Xuchang, Henan province (XC, 34°08′20.4"N, 113°48′34.9"E; northern China), and Qu**g, Yunnan province (QJ, 25°09′40.8"N, 104°01′51.5"E; southwest China). The fertilization trial with maize and wheat/barley rotation was established in spring of 2016 with seven different fertilization treatments as previously described [59]. Finally, an average of 8.6 Gb of clean data was retrieved for each sample. These high-quality reads were assembled using Megahit (v1.2.9) [60], and then were predicted using Prokka (v1.14.5) [61] and clustered with a 0.95 similarity threshold using CD-HIT (v4.8.1) to generate non-redundant gene catalog. The functional profiles including KEGG Orthology (KO), Carbohydrate-Active Enzyme (CAZyome) and Clusters of Orthologous Groups of proteins (COG) of phylloplane microbiomes were determined using eggNOG databases (v5.0) [62], and rarefied based on the lowest reads among all samples (KO 9850; CAZyome 430; and COG 19,710). The Chao1 index of functional diversity was calculated based on rarefied table in QIIME. The linear mixed model (LMM) analysis was performed to identify the major drivers of microbial alpha-diversity using the R package “lme4” [63]. The beta-diversity of both bacterial and fungal communities was assessed by computing weighted UniFrac distance matrices and then ordinated using non-metric multi-dimensional scaling (NMDS). The relative contribution of different biotic and abiotic factors on community dissimilarity was tested with PERMANOVA using the Adonis function (R package “vegan”) [64]. To assess the relative importance of determinism and stochasticity in microbiome assembly, we calculated the beta Nearest Taxon Index (βNTI) using null model (999 randomizations) [65] and defined |βNTI|≥ 2 as dominant deterministic processes and |βNTI|< 2 as dominant stochastic processes [66, 67]. Further, deterministic and stochastic processes were partitioned into five ecological processes based on both βNTI and Bray–Curtis-based Raup-Crick Index (RCBray) values, including heterogeneous selection (βNTI < − 2), homogeneous selection (βNTI > + 2), dispersal limitation (|βNTI|< 2 and RCBray > 0.95), homogenizing dispersal (|βNTI|< 2 and RCBray < – 0.95), and undominated (|βNTI|< 2 and |RCBray|< 0.95) [66, 67]. Microbial interkingdom network analysis at bacterial and fungal genera level was performed using the CoNet [68] in Cytoscape (v3.5) [69] based on Spearman correlation scores (Spearman’s r > 0.7 or r < − 0.7; P < 0.01). Both bacterial and fungal genera present in at least 10 samples were retained for the network analysis [30,31,32,21, 77, 78]. Complementary to the previous finding that host selection via plant compartment niche and host genetics plays a dominant role in sha** plant microbiomes assembly [23, 30, 20, 77, 81]. On the other hand, the effects of plant developmental stage on microbiome in this study included the effects from season-dependent environmental factors like air, dust and rainwater. By using the artificial plants as “background controls” in the field, the impacts of these environmental factors on plant microbiome assembly were discerned in this study. Our results showed that both maize and fake plant phylloplane microbiomes had similar temporal patterns and shared more than one third of ZOTUs at each stage. Further, environmental source (represented by fake plant phylloplane microbiome) contributed an increasing proportion as the source of the maize phylloplane microbiome over the time. These results presented strong field evidence showing that local air, dust and rainwater are the main sources of crop microbiomes in the phyllosphere. These findings significantly advance our knowledge on the source, driving force and potential function of phyllosphere microbiomes, and further corroborated that the phylloplane acts as an important interface between the host, microbes, and the environment [34, 83,84,85]. However, we cannot quantify the specific contribution of each environmental factor like dust and rainwater in the current study, and further research is needed to examine this in the future. Our results also showed that plant developmental stage had significant effects on the rhizosphere and bulk soil microbiomes, though it was much weaker than the site effects, implying that plants also have profound influence on soil microbiomes via the rhizosphere effect [5, 39, 79]. Collectively, by examining the temporal dynamics of bacterial and fungal microbiomes in the soil–plant continuum of maize and fake plant phylloplane in geographically distant sites, this study considerably expanded our knowledge on the succession of plant microbiomes and their potential function under different temporal and spatial scales in field. Bacteria and fungi have coevolved with their host for more than 400 million years and greatly contribute to numerous aspects of plant health and productivity [1, 7, 45]. In this study, bacterial-fungal interkingdom interaction patterns distinctly shifted across three developmental stages. Bacterial community possessed higher alpha-diversity and network connectivity at the seedling stage while fungal diversity was higher at the mature stage. Moreover, bacterial and fungal taxa dominated network hubs at the seedling stage and the mature stage, respectively. These suggested that the host may selectively modulate microbial interactions to meet its requirement during plant growth, as microbial network hubs were supposed to play crucial roles in maintaining plant health and nutrient [41, 44]. In addition, bacterial taxa at the first two stages were better predictors of crop yield while fungal taxa at the mature stage did so. This could be explained by the fact that bacterial community may indirectly affect crop productivity by influencing soil enzyme activities and N availability under different fertilization treatments (Fig. S8; Table S1). Similarly, a recent study has suggested that rice root-inhabiting bacterial microbiota can deeply influence nitrogen-use efficiency of the host plants [86]. Metagenomic analysis further corroborated that maize phylloplane microbiome possessed higher functional diversity at the seedling stage than the other two stages. Importantly, more abundant genes associated with nutrient provision and glycosyl transferases were enriched at the seedling stage while N assimilation- and C degradation-related genes were enriched at two late stages. Based on the limited knowledge on the plant microbiome, it has been proposed that the dynamics of plant microbiome composition are a reflection of the current needs of the host plant [3, 44, 78] and represent the consequence of subtle changes in microbial selection strategy exerted by the host during plant development [1, 24, 78, 87]. Our results therefore supported that bacteria may take a more important ecological role in the plant microbiome and host performance at the early stage, while fungi do so at the late stage. This finding is supported by the null model analysis, which demonstrated the dominant effect of determinism on bacterial community and of stochasticity on fungal community at the seedling stage, but a reverse pattern at the mature stage. In addition, we found that functional gene associated with plant-pathogen interaction (K13457, disease resistance protein RPM1) was significantly enriched at the seedling stage. As the gene is probably derived from the plant genome as the result of biases in plant genome filtering process, the significance of RPM1 enrichment needs further research. The similar bias from shotgun metagenomic sequencing for host microbiomes has also been reported in previous studies [88, 89]. We further found that the negative edges representing bacterial-fungal interkingdom correlations in network increased over the time, implying an increasing competition relationship between bacteria and fungi along plant developmental stages. It was suggested that microbial competitive interaction could positively influence microbiome stability [44, 90, 91]. Our study provided more empirical evidence on this and further supported the argument that the host may facilitate host fitness and plant-microbiome balance by deterministic host selection during plant development. These findings provide new insights into complex interactions among the plant, microbes and the environment and provide essential information for the future development of tools to manipulate crop microbiomes. Our results suggested that the composition and potential functions of plant microbiomes change across plant growth, and more abundant Actinobacteria were observed at the seedling stage than at two late stages in plant compartments. Actinobacteria are well known as antagonistic bacteria excreting antibiotic compounds that provide protection against plant pathogens [92,93,94]. Furthermore, some ZOTUs within families Burkholderiaceae, Streptomycetaceae and Rhizobiaceae were significantly enriched in plant compartment niches at the seedling stage. The members within Burkholderiaceae and Rhizobiaceae are important diazotrophs and plant growth-promoting rhizobacteria (PGPR) [1, 5, 13], and the members within Streptomycetaceae are well-known antibiotic-producing bacteria that are beneficial for plant disease suppression [45, 95, 96]. In addition, bacterial communities in the rhizosphere and bulk soils showed significant correlations with nitrogenase activity across three developmental stages, and the bacterial functional group “nitrite respiration” was identified as the network hubs at the seedling stage. All these suggested that bacterial community takes an ecologically important role in maintaining plant health and nutrient requirement at the early stage. We further found that the fungal classes Sordariomycetes and Dothideomycetes were more sensitive to plant developmental stage. Previous works have shown that Sordariomycetes and Dothideomycetes are the most dominant fungal taxa in soils and plant compartments, respectively, and that class Dothideomycetes comprises a highly diverse range of fungi including endophytes, epiphytes and plant pathogens [46, 97]. In addition, many members within Dothideomycetes are also identified as saprotrophic fungi functioning in wood and leaf-litter decomposition and nutrient cycling [97, 98]. Notably, fungal communities in both fake plant and maize phylloplanes were predominated by Dothideomycetes in two distant study sites across three developmental stages. It was suggested that Dothideomycetes are the dominant fungal taxa of air microbiomes [98]. This indicated that Dothideomycetes in fake plant and maize phylloplanes might be mainly dispersed from air. Furthermore, some fungal ZOTUs affiliating within families Coniothyriaceae, Mycosphaerellaceae and Symmetrosporaceae were identified as network hubs and significantly enriched in plant compartments at the mature stage. Some members of families Coniothyriaceae and Mycosphaerellaceae within Dothideomycetes are important saprobes with cellulose- and carbohydrate-degrading ability [98, 99]. Coincidently, we found that most network hubs in both taxonomic and functional networks of the mature stage belonged to fungal functional group “Saprotroph”. Moreover, fungal communities in the rhizosphere and bulk soils had significant correlations with C cycling-related enzymes like β-glucosidase across three developmental stages. These results suggested that fungal taxa play key roles in regulating plant C cycles like decomposition of plant residues at the late stage. This indicates that crop fungal communities may play an increasing ecological role as the decomposers with the aging of the plant, and the host plant may be passively occupied by saprophytic fungi as the consequence of reduced host immunity. Collectively, our study demonstrates that plant is able to recruit specific microbial taxa with desire functions at different developmental stages. However, the molecular mechanisms governing plant-microbiome interactions during host development and the ecological and biological functions of crop microbiomes in facing climate challenge and achieving sustainable agriculture are not fully understood and need further exploration [11, 100, 101]. By examining the temporal dynamics of bacterial and fungal communities across soils, multiple plant compartments and fake plant phylloplane at two geographically distant sites, this study provides a systematic understanding on the succession of microbiome composition and their potential functions during plant development. Our results demonstrate that plant developmental stage has a much stronger influence on multiple microbial attributes (i.e. alpha-diversity, community structure, determinism/stochasticity patterns and interkingdom networks) in plant compartment niches than in soils, with the strongest effect in the phylloplane. We further found that air is an important source of phylloplane microbiomes, which were strongly co-shaped by plant growth and seasonal environmental factors. Furthermore, we demonstrated that the ecological role of bacterial and fungal community significantly shifts with plant development, along which bacteria take a more important role in maintaining plant health and nutrient requirement at the early stage while fungi take an increasing role in regulating plant C cycles at the late stage. Additionally, we found a dominant effect of determinism on bacterial communities at the early stage and on fungal communities at the late stage in plant compartments. Together these results suggest that the host has a strong selective modulation effect on the composition and potential functions of plant microbiomes during plant development. These findings significantly advance our fundamental understanding of plant-microbiome interactions and provide critical new knowledge for future synthetic community research and the development of microbiome tools to enhance plant protection and agriculture production in a sustainable way.Materials and methods
Field experiment description and sampling
Statistical analysis
The differentiation in ecological roles of bacterial and fungal communities across plant developmental stages
Keystone bacterial and fungal taxa and their ecological functions at different developmental stages
Conclusions
Availability of data and materials
All raw sequencing data have been submitted to the NCBI Sequence Read Archive (SRA) database under the accession numbers PRJNA679910 (16S), PRJNA679909 (ITS), and PRJNA679917 (metagenomics).
References
Martin FM, Uroz S, Barker DG. Ancestral alliances: plant mutualistic symbioses with fungi and bacteria. Science. 2017;356:eaad4501.
Muller DB, Vogel C, Bai Y, Vorholt JA. The plant microbiota: systems-level insights and perspectives. Annu Rev Genet. 2016;50:211–34.
Fitzpatrick CR, Salas-González I, Conway JM, Finkel OM, Gilbert S, Russ D, et al. The plant microbiome: from ecology to reductionism and beyond. Annu Rev Microbiol. 2020;74:81–100.
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A. The importance of the microbiome of the plant holobiont. New Phytol. 2015;206:1196–206.
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol. 2013;11:789–99.
Mendes R, Kruijt M, de Bruijn I, Dekkers E, van der Voort M, Schneider JH, et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 2011;332:1097–100.
Hassani MA, Duran P, Hacquard S. Microbial interactions within the plant holobiont. Microbiome. 2018;6:58.
Fürnkranz M, Wanek W, Richter A, Abell G, Rasche F, Sessitsch A. Nitrogen fixation by phyllosphere bacteria associated with higher plants and their colonizing epiphytes of a tropical lowland rainforest of Costa Rica. ISME J. 2008;2:561–70.
Trivedi P, Leach JE, Tringe SG, Sa T, Singh BK. Plant-microbiome interactions: from community assembly to plant health. Nat Rev Microbiol. 2020;18:607–21.
Sessitsch A, Pfaffenbichler N, Mitter B. Microbiome applications from lab to field: facing complexity. Trends Plant Sci. 2019;24:194–8.
Singh BK, Trivedi P, Egidi E, Macdonald CA, Delgado-Baquerizo M. Crop microbiome and sustainable agriculture. Nat Rev Microbiol. 2020;18:601–2.
Busby PE, Soman C, Wagner MR, Friesen ML, Kremer J, Bennett A, et al. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 2017;15:e2001793.
Haskett TL, Tkacz A, Poole PS. Engineering rhizobacteria for sustainable agriculture. ISME J. 2021;15:949–64.
D’Hondt K, Kostic T, McDowell R, Eudes F, Singh BK. Microbiome innovations for a sustainable future. Nat Microbiol. 2021;6:138–42.
Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–38.
Abdelfattah A, Wisniewski M, Schena L, Tack AJM. Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root. Environ Microbiol. 2021;23:2199–214.
Liu L, Jiang C-Y, Liu X-Y, Wu J-F, Han J-G, Liu S-J. Plant–microbe association for rhizoremediation of chloronitroaromatic pollutants with Comamonas sp. strain CNB-1. Environ Microbiol. 2007;9:465–73.
Vorholt JA, Vogel C, Carlstrom CI, Muller DB. Establishing causality: opportunities of synthetic communities for plant microbiome research. Cell Host Microbe. 2017;22:142–55.
Compant S, Cambon MC, Vacher C, Mitter B, Samad A, Sessitsch A. The plant endosphere world-bacterial life within plants. Environ Microbiol. 2021;23:1812–29.
Harbort CJ, Hashimoto M, Inoue H, Niu Y, Guan R, Rombola AD, et al. Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis. Cell Host Microbe. 2020;28:825-37.e6.
Shakir S, Zaidi SS-e-A, de Vries FT, Mansoor S. Plant genetic networks sha** phyllosphere microbial community. Trends Genet. 2021;37:306–16.
Walters WA, ** Z, Youngblut N, Wallace JG, Sutter J, Zhang W, et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc Natl Acad Sci U S A. 2018;115:7368–73.
diCenzo GC, Checcucci A, Bazzicalupo M, Mengoni A, Viti C, Dziewit L, et al. Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti. Nat Commun. 2016;7:12219.
Viviane C, Francisco DA, Víctor JC, Jos MR. Ecology and evolution of plant microbiomes. Annu Rev Microbiol. 2019;73:69–88.
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90.
Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–5.
Thiergart T, Duran P, Ellis T, Vannier N, Garrido-Oter R, Kemen E, et al. Root microbiota assembly and adaptive differentiation among European Arabidopsis populations. Nat Ecol Evol. 2020;4:122–31.
Hartman K, van der Heijden MGA, Wittwer RA, Banerjee S, Walser JC, Schlaeppi K. Crop** practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome. 2018;6:14.
Schmidt JE, Kent AD, Brisson VL, Gaudin ACM. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome. 2019;7:146.
Cregger MA, Veach AM, Yang ZK, Crouch MJ, Vilgalys R, Tuskan GA, et al. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome. 2018;6:31.
Beckers B, Op De Beeck M, Weyens N, Boerjan W, Vangronsveld J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome. 2017;5:25.
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A. 2015;112:E911–20.
**ong C, Zhu YG, Wang JT, Singh BK, Han LL, Shen JP, et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021;229:1091–104.
Lindow SE, Brandl MT. Microbiology of the phyllosphere. Appl Environ Microbiol. 2003;69:1875–83.
Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, et al. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat Commun. 2020;11:1–14.
Grady KL, Sorensen JW, Stopnisek N, Guittar J, Shade A. Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat Commun. 2019;10:4135.
Zhang JY, Zhang N, Liu YX, Zhang XN, Hu B, Qin Y, et al. Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci China Life Sci. 2018;61:613–21.
Chen S, Waghmode TR, Sun R, Kuramae EE, Hu C, Liu B. Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization. Microbiome. 2019;7:136.
Zhao ML, Zhao J, Yuan J, Hale L, Wen T, Huang QW, et al. Root exudates drive soil-microbe-nutrient feedbacks in response to plant growth. Plant Cell Environ. 2021;44:613–28.
Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:e1002352.
van der Heijden MGA, Hartmann M. Networking in the plant microbiome. PLoS Biol. 2016;14:e1002378.
Barberan A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51.
Toju H, Tanabe AS, Sato H. Network hubs in root-associated fungal metacommunities. Microbiome. 2018;6:116.
Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–6.
Duran P, Thiergart T, Garrido-Oter R, Agler M, Kemen E, Schulze-Lefert P, et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell. 2018;175:973-83.e14.
**ong C, He JZ, Singh BK, Zhu YG, Wang JT, Li PP, et al. Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ Microbiol. 2021;23:1907–24.
Marx MC, Wood M, Jarvis SC. A microplate fluorimetric assay for the study of enzyme diversity in soils. Soil Biol Biochem. 2001;33:1633–40.
Tabatabai M. Soil enzymes. In: R.W. Weaver SA, P. Bottomley, D. Bezdicek, S. Smith, A. Tabatabai and A. Wollum, editors. Methods of soil analysis. Madison: Soil Science Society of America; 1994. p. 775–833.
Kembel SW, O’Connor TK, Arnold HK, Hubbell SP, Wright SJ, Green JL. Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc Natl Acad Sci U S A. 2014;111:13715–20.
Ihrmark K, Bodeker IT, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, et al. New primers to amplify the fungal ITS2 region-evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol. 2012;82:666–77.
White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols. San Diego: Academic Press; 1990. p. 315–22.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016;081257. https://doi.org/10.1101/081257.
Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–7.
Nguyen NH, Song ZW, Bates ST, Branco S, Tedersoo L, Menke J, et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016;20:241–8.
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of highthroughput community sequencing data. Nat Methods. 2010;7:335–6.
Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–2.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018;47:D309–14.
Nakagawa S, Schielzeth H, O’Hara RB. A general and simple method for obtaining r2 from generalized linear mixed-effects models. Methods Ecol Evol. 2013;4:133–42.
Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, et al. The vegan package. Community Ecol Package. 2007;10:631–7.
Stegen JC, Lin XJ, Fredrickson JK, Chen XY, Kennedy DW, Murray CJ, et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013;7:2069–79.
Jiao S, Yang Y, Xu Y, Zhang J, Lu Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020;14:202–16.
Zhou J, Deng Y, Zhang P, Xue K, Liang Y, Van Nostrand JD, et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci U S A. 2014;111:E836–45.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8:e1002606.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. Icwsm. 2009;8:361–2.
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Methods. 2011;8:761–3.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Liaw A, Wiener M. Classification and regression by randomForest. R news. 2002;2:18–22.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Singh BK, Trivedi P. Microbiome and the future for food and nutrient security. Microb biotechnol. 2017;10:50–3.
Chaparro JM, Badri DV, Vivanco JM. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2013;8:790–803.
Huang AC, Jiang T, Liu YX, Bai YC, Reed J, Qu B, et al. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science. 2019;364:eaau6389.
Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. The evolution of the host microbiome as an ecosystem on a leash. Nature. 2017;548:43–51.
Sasse J, Martinoia E, Northen T. Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci. 2018;23:25–41.
Guttman DS, McHardy AC, Schulze-Lefert P. Microbial genome-enabled insights into plant-microorganism interactions. Nat Rev Genet. 2014;15:797–813.
Kudjordjie EN, Sapkota R, Steffensen SK, Fomsgaard IS, Nicolaisen M. Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome. 2019;7:59.
Hu J, Wei Z, Kowalchuk GA, Xu Y, Shen Q, Jousset A. Rhizosphere microbiome functional diversity and pathogen invasion resistance build up during plant development. Environ Microbiol. 2020;22:5005–18.
Vacher C, Hampe A, Porté AJ, Sauer U, Compant S, Morris CE. The phyllosphere: microbial jungle at the plant–climate interface. Annu Rev Ecol Evol S. 2016;47:1–24.
Remus-Emsermann MNP, Schlechter RO. Phyllosphere microbiology: at the interface between microbial individuals and the plant host. New Phytol. 2018;218:1327–33.
Vorholt JA. Microbial life in the phyllosphere. Nat Rev Microbiol. 2012;10:828–40.
Zhang J, Liu YX, Zhang N, Hu B, ** T, Xu H, et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol. 2019;37:676–84.
Finkel OM, Salas-Gonzalez I, Castrillo G, Conway JM, Law TF, Teixeira PJPL, et al. A single bacterial genus maintains root growth in a complex microbiome. Nature. 2020;587:103–8.
New FN, Brito IL. What is metagenomics teaching us, and what is missed? Annu Rev Microbiol. 2020;74:117–35.
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35:833–44.
Santolini M, Barabasi AL. Predicting perturbation patterns from the topology of biological networks. Proc Natl Acad Sci U S A. 2018;115:E6375–83.
Banerjee S, Walder F, Buchi L, Meyer M, Held AY, Gattinger A, et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019;13:1722–36.
Alvarez-Perez JM, Gonzalez-Garcia S, Cobos R, Olego MA, Ibanez A, Diez-Galan A, et al. Use of endophytic and rhizosphere Actinobacteria from grapevine plants to reduce nursery fungal graft infections that lead to young grapevine decline. Appl Environ Microbiol. 2017;83:e01564-e1617.
Lee SM, Kong HG, Song GC, Ryu CM. Disruption of Firmicutes and Actinobacteria abundance in tomato rhizosphere causes the incidence of bacterial wilt disease. ISME J. 2021;15:330–47.
Viaene T, Langendries S, Beirinckx S, Maes M, Goormachtig S. Streptomyces as a plant’s best friend? FEMS Microbiol Ecol. 2016;92:fiw119.
Conn VM, Walker AR, Franco CMM. Endophytic actinobacteria induce defense pathways in Arabidopsis thaliana. Mol Plant Microbe Interact. 2008;21:208–18.
Trivedi P, Delgado-Baquerizo M, Trivedi C, Hamonts K, Anderson IC, Singh BK. Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil Biol Biochem. 2017;111:10–4.
Hyde KD, Jones EG, Liu JK, Ariyawansa H, Boehm E, Boonmee S, et al. Families of Dothideomycetes. Fungal Diversity. 2013;63:1–313.
Adams RI, Miletto M, Taylor JW, Bruns TD. Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013;7:1262–73.
Egidi E, Delgado-Baquerizo M, Plett JM, Wang J, Eldridge DJ, Bardgett RD, et al. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat Commun. 2019;10:1–9.
Cheng YT, Zhang L, He SY. Plant-microbe interactions facing environmental challenge. Cell Host Microbe. 2019;26:183–92.
Toju H, Peay KG, Yamamichi M, Narisawa K, Hiruma K, Naito K, et al. Core microbiomes for sustainable agroecosystems. Nat Plants. 2018;4:247–57.
Acknowledgements
We would like to thank Prof. Yong-Guan Zhu, Dr. Hang-Wei Hu and Dr. Yin Chen for their suggestions and comments on experiment design and manuscript writing, and Mr. Qin-Bing Zhang and Hao-Tian Tong for their cooperation and assistance in field management and sampling. We also thank Mr. Feng Dai for supplying the nitrification inhibitor chlorinated pyridine (CP).
Funding
This work was financially supported by the National Key R&D Program (2017YFD0200600) and the Strategic Priority Research Program (B) of Chinese Academy of Sciences (XDB15020200). Research on plant and soil microbiomes in BKS lab is supported by the Australian Research Council (DP170103628; DP190103714). Li-Mei Zhang was supported by the Youth Innovation Promotion Association, Chinese Academy of Sciences (Y201615).
Author information
Authors and Affiliations
Contributions
LMZ, JZH, and CX designed the study. LMZ, SYL, GZM, LHW, YLH and PPL managed the field trial stations. CX, CFW and AHG collected samples. CX conducted the laboratory analyses. CX, JTW, BS and LMZ performed the data processes. CX, LMZ, BS, JTW and JZH wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
**ong, C., Singh, B.K., He, JZ. et al. Plant developmental stage drives the differentiation in ecological role of the maize microbiome. Microbiome 9, 171 (2021). https://doi.org/10.1186/s40168-021-01118-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01118-6