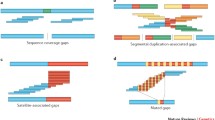
Variant discovery from assembly-versus-assembly alignment
Our approach starts with assembly-versus-assembly alignment, for which we use the LAST aligner [16] with the application of a split-alignment algorithm (Martin Frith, personal communication). In the assembly-versus-assembly alignment, we transverse each scaffold from 5’ to 3’ and record variants when mismatches, small insertions or deletions (indels) or other more complex forms of genome rearrangements are observed in one alignment block, or when breakpoints between two linear alignment blocks occur (Fig. 1a). We categorize the variations between the reference and the individual de novo assembly into ‘SNP’, ‘deletion’, ‘insertion’, ‘inversion’, ‘simultaneous gap’, or ‘intra- and inter-chromosomal translocation’, whereas the ones that cannot be characterized are categorized as ‘no solution’. We group the unaligned sequences in the de novo assembly as ‘clipped sequences’ or ‘nomadic sequences’; these are novel sequence candidates, but could also be due to contamination, assembly errors or other artifacts. The reference regions that are not covered by the de novo assembly are categorized as ‘inter-scaffold gaps’ or ‘intra-scaffold gaps’, and they are often associated with large repetitive sequences in the human genome or result from insufficient sequencing depth.
Around the breakpoints of the structural variants, we use an align-gap-excise alignment algorithm [17] to perform local realignment (Fig. 1a). In this process, all the variants are left-shifted and the representations of complex variants are unified, which facilitates population genetics studies of variation [18]. Subsequently, we combine all the variants from different de novo genome assemblies and store them in standard Variant Call Format (VCF) in accordance with the conventions of the 1000 Genomes Project (Fig. 1b) [4]. When performing this step of the approach, we recommend including the publicly available de novo genome assemblies from the same population (termed prior de novo assemblies in Fig. 1b) to increase the discovery power and provide prior information for the subsequent variant score recalibration process.
Individual genoty**
We genotype the structural variants using a linear-constrained Gaussian mixture model with three states, AA, AR and RR, assuming that a reference allele (R) and an alternative allele (A) are segregating in the human population. The Gaussian mixture process models the density of the two-dimensional variables that record the normalized counts of reads that support the reference allele (R intensity) and the alternative allele (A intensity). Both intensities are obtained by realigning reads against the two alleles (Fig. 1c).
We constrain the centres of the three genotype states on the basis of the expected A and R intensities for each state and approximate the weight of the Gaussian mixture model by the proportion of individuals in the population with a certain genotype. We optimize the parameters in the Gaussian mixture model using an expectation-maximization (EM) algorithm with linear constraints. With the expected weight, centres and corresponding standard deviations obtained from the training process, we calculate the genotype likelihood, decide the genotype and estimate the genotype quality for each individual (see Additional file 2: Supplementary Methods for details).
In formulation, for a particular variant in the individual i, the genotype posterior probability of a particular genotype j is computed as follows:
$$ P\left({G}_{ij}\Big|{d}_i\right) = \frac{w_jN\left({d}_i\Big|{\mu}_j,\ {\Sigma}_j\right)}{{\displaystyle {\sum}_{j=1}^K}{w}_jN\left({d}_i\Big|{\mu}_j,\ {\Sigma}_j\right)} $$
(1)
G
ij
represents the assumed genotype j for the individual i; d
i
represents the two-dimension vector that composes R intensity (the count of the reads uniquely aligned to the reference allele R divided by the total depth) and A intensity (the count of the reads uniquely aligned to the alternative allele A divided by the total depth) for the individual i; w
j
indicates the proportion of individuals that have genotype state j; μ
j
is the expected value of mean of d
i
given genotype state j; Σ
j
is the expected value of standard deviation of d
i
given genotype state j. N(d
i
|μ
j
, Σ
j
) is the probability of observing d
i
providing the Gaussian mixture model with mean and standard deviation μ
j
and Σ
j
. K refers to the total number of genotype states and is constantly 3 because our model only considers bi-allelic loci so far. We will release a new model that accommodates a multi-allele situation in AsmVar version 2.0.
The likelihood of observing d
i
given a particular genotype G
ij
is:
$$ P\left({d}_i\Big|{G}_{ij}\right) = {w}_jN\left({d}_i\Big|{\mu}_j,\ {\varSigma}_j\right) $$
(2)
Supposing all the individuals are unrelated to each other, the log likelihood function is constructed as follows:
$$ ln\ P\left(D\Big|w,\mu, \varSigma \right) = \ln \Big({\displaystyle {\displaystyle {\displaystyle {\sum}_{j=1}^K\left({\displaystyle {\displaystyle {\sum}_{i=1}^N}}\ {w}_jN\left({d}_i\Big|{\mu}_j,\ {\varSigma}_j\right)\ \right)}}} $$
(3)
w, μ, Σ are optimized using an EM algorithm with linear constraints. D refers to the set of all the observed data d
i
. The initial values for μ are centered around [0.001,0.001], [0.5, 0.5] and [1.0, 1.0], corresponding to the homozygous reference allele (RR), heterozygous variants (RA) and homozygous variants (AA) genotype states, respectively. These values are multiplied by a scaling factor m that ranges from 0.8 to 1.2 with interval 0.1 and therefore there will be five rounds of training. The best m is selected on the basis of the bias from a set of linear constraints and the Mendelian errors (see Additional file 2: Supplementary Methods for details). The initial value for the vector w, i.e. genotype frequency for three genotype states is [1/3, 1/3, 1/3].
The genotype of the individual (G
ij
) is selected as the one out of the three that has the highest posterior probability.
The Phred-scale genotype quality score (GQi) is estimated by:
$$ G{Q}_i = -10*\ \log 10\left(1-\frac{P\left(G{T}_i\Big|d\right)}{{\displaystyle {\sum}_{j=1}^K}P\left(G{T}_i\Big|d\right)}\right) $$
(4)
Variant quality score recalibration
Similar to the approach implemented in GATK [2], we apply a Bayesian Gaussian mixture model to the raw variant calls to assign a quality score and classify the variants as PASS and FALSE. This is a classification process guided by a positive training set, a negative training set, a set of technical features and, optimally, an independent validation set (Fig. 1d).
The positive and negative sets consist of true positive and true negative variants with additional experimental or computational evidence. We offer the users options to include their own training and validation sets. The positive sets can be the variants that are known to be polymorphic, variants independently assembled in more than one individual (double-hit events), variants that have additional computational evidence (such as the ones that are called with other software tools) or ideally variants that have been experimentally validated. The negative sets are variants known to be artifacts.
Three types of false-positive sources exist: assembly error, global alignment errors and local alignment artifacts. AsmVar captures nine metrics associated with these sources of error, including: the local assembly gap ratio; the depth of the reads that support the alternative allele; the depth of the reads that neither support the reference allele nor the alternative allele; the misalignment probability and the alignment score of the scaffolds that carry the structural variants; the local sequence identity; the position of the variants in the scaffold; and the proper aligned read ratio; and the improper aligned read ratio in the short-read versus the reference alignment (see Additional file 2: Figure S3). The users can specify all of these features or only a few selected features in the training.
We fit the quantitative measurements of a selected set of these technical features into the Gaussian mixture model and compute the log odds ratio of the likelihood that the observed variant arises from the positive training model versus the likelihood that it comes from the negative training model.
Below is the formulization of the recalibration process:
p01 and p02 are the prior probability for the variants conditioned on being positive and negative, respectively. We assign known variants with higher prior probability of being positive compared to that of the novel ones. m is the number of the cluster in the guassian mixture model ranging from 1 to the maximum number 8 by default. w indicates the size of a certain center provided m. x is a vector that records the distribution of the features.
$$ P\left(x\Big|{G}_{positive}\right) = {p}_{01}(x){\displaystyle {\sum}_{i=1}^m{w}_i}N\left(x\Big|{\mu}_i,{{\displaystyle \sum}}_i\right) $$
(5)
$$ P\left(X\Big|{G}_{Negative}\right) = {p}_{02}(x){\displaystyle {\sum}_{j=1}^n{w}_j}N\left(x\Big|{\mu}_j,{{\displaystyle \sum}}_j\right) $$
(6)
$$ {p}_{01}(x)=\left\{\begin{array}{c}\hfill 0.6,\kern0.5em x\ is\ known\ variant\hfill \\ {}\hfill \kern1.25em 0.4,\kern3.75em Otherwise\kern2.75em \hfill \end{array}\right. $$
(7)
$$ {p}_{02}(x)=\left\{\begin{array}{c}\hfill 0.4,\kern0.5em x\ is\ known\ variant\hfill \\ {}\hfill \kern1.25em 0.6,\kern3.75em Otherwise\kern2.75em \hfill \end{array}\right. $$
(8)
$$ Score(x)=- \lg \left(1-P\left(x\Big|{G}_{positive}\right)\right)+ lg\left(1-P\left(x\Big|{G}_{negative}\right)\right) $$
(9)
The quality score threshold is determined so as to maximize the area under the receiver operating characteristic (ROC) curve (AUC), where we keep most of the known positive variants while minimizing the inclusion of the known negative variants. It is better if the known positive and negative training variants (validation) are independent sets from the validation sets. However, when lacking such independent sets, the users can also use the option -cv in AsmVar to invoke the cross validation module, which uses the training set to assess the error rate.
Since excessive heterozygosity and homozygosity are good indicators of genoty** errors [7], we also apply the inbreeding coefficient to filter the loci with excessive heterozygosity or homozygosity (6). According to the latest investigations of artifacts in variant calling from high-coverage samples [7] and our own observations, excessive heterozygosity is relevant to the existence of large segmental duplications, whereas excessive homozygosity can derive from the assembly errors of the human genome reference or from cryptic systematic errors during data processing and variation calling.
The inbreeding coefficient (F) is computed as below:
$$ F = 1.0 - \left({N}_{\mathrm{het}}/\ \left(\ 2.0\ *p*q*N\right)\ \right) $$
(10)
Where p and q are the sample allele frequencies (only the 20 parents are considered in our study of ten Danish trios) of the reference and alternative alleles, respectively.
N refers to the total number of unrelated individuals in a population.
Nhet refers to the total number of unrelated individuals (N) that are heterozygous.
By default, AsmVar removes variants with an inbreeding coefficient < -0.4 or >0.7. The threshold for inbreeding coefficient is determined based on the basis of its distribution (see Additional file 2: Figure S12), taking the GATK experience into consideration [2].
Characterization of the ancestral state of the structural variants
After obtaining the structural variants present in the de novo genome assemblies, we annotate the ancestral allele state of a structural variant by comparing the identity and the aligned ratio of the reference allele and the alternative allele to the orthologous region in an outgroup genome, such as a primate genome when analyzing human sequences (Fig. 1e). By default, AsmVar uses four primate genomes (Chimpanzee panTro4, Orangutan ponAbe2, Gorilla gorGor3, Macaque rheMac3) as the outgroup genomes for comparisons. The allele that has substantially higher identity and aligned ratio to the orthologous region of the outgroup genome is identified as the ancestral allele.
We first construct the reference and the alternative alleles taking the flanking 500 bp around the variant region into account. We align both the reference and the alternative alleles to the genomes of the four primates using LAST [16] and measure the similarity using the identity and aligned ratio from the alignment. We categorize the variants as: ‘NONE’, when both the reference and the alternative alleles cannot be aligned to any of the primate genomes; ‘NA’, when both the reference and the alternative alleles can be aligned to one of the primate genomes but has less than 95 % identity and 95 % aligned ratio for all four primates; ‘Common’, when both the reference and the alternative alleles have greater than 95 % identity and aligned ratio for all four primate genomes; ‘Deletion’, when the longer allele has greater than 95 % identity and aligned ratio for any of the primate genomes and the shorter allele has less than 95 % identity and aligned ratio for any of the primate genomes; ‘Insertion’, when the longer allele has greater than 95 % identity and aligned ratio for any of the primate genomes and the shorter allele has less than 95 % identity and aligned ratio for any of the primate genomes; and ‘Conflict’, when the ‘Insertion’ and ‘Deletion’ judgment is different between different primate genomes.
Finally, we rectify the types of variation on the basis of the ancestral allele state. For example, if the assembly-versus-reference alignment suggests an insertion, but ancestral state analysis indicates that the assembly allele is the ancestral allele, we eventually annotate this variant as a deletion instead (Fig. 1e).
Characterization of formation mechanisms of the structural variants
We characterize the formation mechanism of a variant according to the pattern of repeats in and around the variant sequence using a classification scheme similar to the BreakSeq method [19] and the 1000 Genomes Project approach [20]. Briefly, we align the variant allele sequences to RepBase using RepeatMasker [21] and perform reciprocal alignment between the left and right breakpoint sequences using BLASTn [22] (Fig. 1e; Methods). The assembly alleles that show substantial similarity with simple repeats or mobile element sequences in RepBase are annotated as variable number of tandem repeats (VNTR) or transposable element insertion (TEI), respectively. The variants that have more than 85 % identity between the two breakpoints are annotated as non-allelic homologous recombination (NAHR). Variations that contain short tracts of identical sequences around the breakpoint (micro-homology phenomena) are annotated as non-homologous rearrangements (NHR). In addition, if the full variant sequence is completely identical to the 3’ sequence of the right breakpoint, it is annotated as copy count change (CCC), which mainly derives from DNA polymerase slippage [20].
Novel sequence
In addition to structural variants, we identify novel sequence insertions and novel sequences that are not well aligned to the consensus human genome reference but have high similarity to other human and primate genomes (identity ≥0.95 and align ratio ≥0.95) (Fig. 1f; see Additional file 2, Supplementary Methods for details). We analyse the distribution, ancestral state and mechanism of formation of all novel sequence and link the novel sequences to the closest sequences from known de novo assemblies.
Scalability
AsmVar is highly efficient and currently takes only approximately 16 h to discover, genotype and characterize the structural variants and novel sequences from a de novo assembly using 8 CPU cores and 64 GB of memory (see Additional file 3: Table S2).
Conventions and graphical presentation
To facilitate downstream analysis and research communication, we record the structural variants in a standard VCF [23], according to the 1000 Genomes Project convention. AsmVar also summarizes the types, size spectrum, ancestral state and formation mechanism of the structural variants and novel sequences from the investigated samples graphically in demo plots.
A complete description of the AsmVar approach is provided in Additional file 2: Supplementary Methods.
Discovery and genoty** of structural variants from 37 human de novo genome assemblies
We show the utility of the AsmVar strategy by applying this tool to systematically investigate the structural variants and novel sequence in the currently available de novo assemblies of the human genome. By 31 July 2014, 37 human de novo assemblies are accessible to us, which include the ten Danish trios from the Genome Denmark consortium [14] and another seven de novo assemblies. Detailed information about the 37 de novo assemblies is listed in Additional file 1: Table S1. We present the results in a series of demo plots generated by the AsmVar package.
Using the AsmVar strategy, we initially identify a total of 8,609,194 raw non-SNP variants and subsequently assign genotype likelihoods, genotype and genotype quality to each individual. As a positive control set, we randomly select a subset of 626,028 double-hit exact breakpoint structural variants that are independently assembled from more than two individuals (see Additional file 2: Figure S2). We then quantify the variant quality score in the recalibration module l (see Additional file 2: Figure S3). Finally we obtain 3,176,200 structural variants from the 37 de novo assemblies, with lengths that range from 1 bp to 50 kbp; approximately 93 % of the positive training variants can be recovered and the false-positive rate is approximately 0.7 % (see Additional file 2: Figure S4).
As shown in Fig. 2, our approach reveals a variety of structural variants with nucleotide resolution, which include 1,194,473 deletions, 1,151,871 insertions, 14,745 block substitutions, 587,143 length-asymmetric replacements, 171 inversions and 223,477 translocations. The variants range from 1 bp to 100 kbp, with peaks around 300 bp and 6 kbp, which correspond to transposition events that took place in the evolution of human populations (Fig. 3a). The individual load and size spectrum of the structural variants approximate those reported by the HuRef genome investigation [24], but these data have been consistently missed in genome analyses in which re-sequencing-based approaches were used. The latter mainly restricts in deletion investigations and displays substantial bias over size spectrum and resolution (see Additional file 2: Figure S5) [4, 5].
Benchmarking the sensitivity and specificity of structural variant genoty** by AsmVar
We benchmark the AsmVar approach using both computational and experimental evidence. As 51.14 % of the structural variants identified by AsmVar (N = 1,624,308) are novel, that is, not present in the current dbVar database, we perform computational validation of the novel callset. By observing a random selection of 600,000 of the novel structural variants, we discover that the normalized read intensity is systematically stronger for the alternative allele than for the reference allele (see Additional file 2: Figure S6). This finding suggests that most of the novel structural variants are true polymorphisms within the human population.
We subsequently evaluate the structural variant genoty** performance of AsmVar using population metrics including family relatedness and the Mendelian error rate. Those metrics are computed using the PLINK software [25]. The probability of identity by descent being equal to 1 (IBD1) for the parent-offspring genomes varies from 0.02 to 0.14 for deletions and 0.10 to 0.19 for insertions, whereas the probability of pairwise IBD0 for unrelated individuals approximates zero (see Additional file 2: Figure S7). The Mendelian error rate ranges from 0.01 to 0.21 for deletions and 0.03 to 0.10 for insertions (see Additional file 2: Figure S8). Based on these metrics, we estimate that the genoty** error for AsmVar calls is approximately 2 % to 20 %. Although the performance of AsmVar for structural variant genoty** is not as good as that for GATK SNP identification, the genoty** accuracy of AsmVar substantially exceeds that of the most widely used software for structural variation genoty**, GenomeStrip [6], which was the structural variation caller and genotyper adopted in the 1000 Genomes Project (see Additional file 2: Figure S7 and Figure S8).
Furthermore, we benchmark the performance of AsmVar using two datasets for which experimental evidence exists. First, as NA12878, which is included in our study, is a well-studied individual genome, we benchmark the sensitivity of AsmVar by comparing the NA12878 AsmVar non-reference genotype calls to the 21415 dbVar structural variation records for this individual [5]. These structural variants include 18,108 deletions, 294 insertions, 491 duplications and 39 inversions that are >50 bp and were validated by different experimental approaches. Also, there were 2050 deletions, 152 insertions, 244 duplications and 37 inversions that failed experimental validation.
Among the validated structural variants, 3738 are missed by AsmVar without enrichment of a certain size spectrum (see Additional file 4: Table S3). Therefore, the overall false-negative rate of AsmVar is approximately 20.1 %. Manual investigation into these missing calls suggests three main reasons for false-negative calls: 1) assembly gaps due to insufficient coverage; 2) assembly gaps derived from long repetitive sequences; and 3) assembly errors probably result from underlying complex genomic sequences.
AsmVar calls none of the 2483 variants from the NA12878 dbVar dataset that failed validation. However, as the true number of variants present in NA12878 is not available at the moment based on our observations of the Illumina Platinum Genomes and Genome In A Bottle datasets [18], we are not able to unbiasedly assess the false-positive rate of AsmVar using the NA12878 public data. In addition, as genotype information about structural variants in the NA12878 dbVar records is not available, we cannot benchmark the genoty** accuracy of AsmVar using the dbVar information.
To further assess the specificity of AsmVar in structural variation discovery, we randomly select one Danish trio from the Genome Denmark consortium and validate 272 novel structural variants with a range of different sizes (≥50 bp) and formation mechanisms using the Sanger sequencing technology [14]. We successfully assay 68 structural variants, and from this analysis we estimate that the overall false-discovery rate of AsmVar for structural variants is 7.4 % (5/68, 95 % confidence interval = 0.03-0. 16) (see Additional file 5: Table S4). For the remaining 204 loci, 158 are not successfully assayed because of failure in primer design and 46 are not successfully assayed because of other experimental problems, such as the failure of the PCR or sequencing.
The validation of structural variation remains a challenge. The experimental failure rate is high, probably because most of the structural variants occur in repetitive sequences of DNA. We therefore include in the AsmVar package an extension script to plot out the proper and the improper read coverage at and around the loci in which structural variation was identified (see Additional file 2: Supplementary Methods, for definition of proper and improper reads; see also Additional file 2: Figure S9). Manual inspection indicates that the false-positive rates for the two categories of failure attempts are 6.5 % and 8.2 %, respectively. Owing to the limited number of validation loci available for each size band or for each type of formation mechanism, we cannot correlate the false-discovery rate with the size spectrum and the formation mechanism of the variants with high confidence.
The ancestral state of the structural variants
One characteristic of the variants in AsmVar is that their sequences are available, which is the precondition to define the ancestral state of a variant. To obtain insight into the evolutionary origin of the structural variants obtained from the 37 human de novo assemblies that were included in this study, we apply AsmVar to analyze the ancestral state of the variants according to the size spectrum. We summarize the AsmVar results using the demo plot functionality (Fig. 3b). Owing to the lower quality of some of the primate genomes when compared with that of the human de novo assemblies, we cannot characterize the ancestral state of 51.2 % of the variants. By comparing the human datasets to the outgroup genomes, we discover that 9 % of the insertions in the de novo assemblies show higher similarity to the outgroup genomes than to the human reference genome and are indeed evolutionally deletion events in the first beginning. This observation also highlights the incompleteness of the consensus human genome reference (Fig. 3b). Conversely, we discover that 28 % of the classified deletions are instead insertion events. Consistent with the molecular level understanding, the deletions that have arisen owing to TEI mechanisms tend to be insertions in the historical course (Fig. 3b). Our approach reveals similar patterns of distribution of ancestral states among structural variants than those reported in previous population-scale investigations in which a set of large deletions and a very limited number of tandem duplications were analyzed [5].
The formation mechanism of the structural variants
Nucleotide resolution of the structural variants identified using AsmVar enables the characterization of their mechanisms of formation. We classify the mechanisms of formation of the structural variants into VNTR, TEI, NAHR, NHR and CCC, i.e. copy number changes derived from a DNA polymerase slippage process across the size spectrum (Fig. 3c). Our approach demonstrates a symmetric view of mechanisms distribution corresponding to our molecular level understandings. Most of the 1–10 bp insertions and deletions have exact copy number changes that are relevant to DNA polymerase slippage. The 300 bp and the 6 kbp variants are enriched in TEI and the larger variations (>1,000 bp) arise from NAHR and NHR, whereas the smaller ones are enriched in VNTR [15]. Most of the TEI-derived deletions indeed have insertions as the ancestral state. These observations follow our biological intuition, which indirectly proves the robustness of our approach.
Novel sequence
In addition to structural variants, we identify 9 million base pairs of novel sequences (>100 bp), on average, per individual that are not present in the human genome reference sequence, as shown in the AsmVar demo plot (Fig. 4).
We divide the novel sequences into novel sequence insertions and nomadic novel sequences (Fig. 1g). We first investigate the ancestral state, the formation mechanism and chromosomal distribution of the novel sequence insertions. 90 % of the novel inserted sequences show higher similarity to the outgroup primate genomes compared to the human reference genome. Therefore, we observe a higher number of deletions than insertions in the ancestral state analysis, which correspond to NHR and NAHR molecular mechanisms of origin [19] (Fig. 4a). The novel sequence insertions are distributed across the whole human genome, affecting the structure of 71 genes. We randomly select 18 large novel sequence insertions (≥1 kbp) and apply quantitative PCR (qPCR) to validate their existence. Manual observation of the electrophoretic band validates all of these insertions (see Additional file 6: Table S5). However, AsmVar predicts the insertion length incorrectly for one locus.
We subsequently learn about the un-localized novel sequences identified by AsmVar by linking each of the sequences of one individual to their closest neighbour (Fig. 4b). We notice that CHM assembly contains a very limited number of novel sequences and confirm that this assembly is a reference-guided de novo assembly. This finding also highlights a bias of re-sequencing-based approaches for investigation of genome variation. Except for CHM genome assembly, we observe that the proportion of nomadic sequences decreases as the quality of the de novo assembly increases. We reason that a high-quality de novo assembly contains novel sequences that cannot be captured by de novo assemblies with lower quality. When investigating the closest relatives of the novel sequences, we observe a consistent ranking of proportion from HuRef to YH and NA12878, which corresponds to the quality of these de novo assemblies. These observations indicate that obtaining a comprehensive profile of the variations present in a human genome relies on high-quality de novo assemblies (Fig. 4b).