
Abstract
Single-cell RNA sequencing (scRNA-seq) is a tool for studying gene expression at the single-cell level that has been widely used due to its unprecedented high resolution. In the present review, we outline the preparation process and sequencing platforms for the scRNA-seq analysis of solid tumor specimens and discuss the main steps and methods used during data analysis, including quality control, batch-effect correction, normalization, cell cycle phase assignment, clustering, cell trajectory and pseudo-time reconstruction, differential expression analysis and gene set enrichment analysis, as well as gene regulatory network inference. Traditional bulk RNA sequencing does not address the heterogeneity within and between tumors, and since the development of the first scRNA-seq technique, this approach has been widely used in cancer research to better understand cancer cell biology and pathogenetic mechanisms. ScRNA-seq has been of great significance for the development of targeted therapy and immunotherapy. In the second part of this review, we focus on the application of scRNA-seq in solid tumors, and summarize the findings and achievements in tumor research afforded by its use. ScRNA-seq holds promise for improving our understanding of the molecular characteristics of cancer, and potentially contributing to improved diagnosis, prognosis, and therapeutics.
Similar content being viewed by others
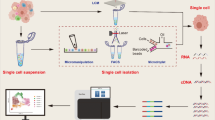


Background
Tumors are generally considered to be of monoclonal origin, with mutations facilitating the expansion of a malignant cell in the body to visible tumor tissue, as well as from carcinoma in situ to metastatic carcinoma. Notably, patient-derived tumor tissue includes cancer cells as well as other cell types, such as infiltrating immune cells and fibroblasts. Therefore, the use of traditional bulk RNA sequencing technology can only permit an average qualitative characterization of such highly complex tissue, representing the average of cancer cells and non-cancer cells. Although bulk RNA approaches have made many invaluable contributions to medical science [1], such approaches have ignored the distinct phenotypical and functional traits of single cells within tumor samples.
Since the first single-cell RNA sequencing (scRNA-seq) study was published in 2009 [2], various commercial platforms and methods have been developed for scRNA-seq. ScRNA-seq is a technique allowing for the study of tissues at single-cell resolution. Many researchers within the life sciences employ scRNA-seq for the investigation of diverse biological functions. In particular, great achievements have been made in tumor research using scRNA-seq technology. The single-cell resolution afforded by scRNA-seq enables direct measurement of the transcriptional output of cells from tumor samples [3], comparison of differences between the transcriptomes of various cells, identification of rare cell subpopulations, such as heterogeneous tumor subpopulations [4], or the revelation of differences between stimulated dendritic cells [5], in turn providing unprecedented insights that have contributed to the development of cancer therapy.
In the present review, we introduce methods for the preparation of solid tumor samples, related scRNA-seq platforms, the analysis of scRNA-seq data, and achievements in tumor research facilitated by the use of scRNA-seq.
Single-cell RNA sequencing
To carry out scRNA-seq, solid tumor samples first need to be processed to effectively isolate viable single cells from the tissue of interest [6]. Thereafter, the single cells are lysed to obtain RNA, which is reverse transcribed into cDNA and then amplified to construct a sequencing library. The most suitable sequencing instrument must be selected based on the experimental scheme and research objectives. After sequencing, the data need to be correctly analyzed to reveal new findings.
Preparation of solid tumor specimens
Many published articles on the use of scRNA-seq in cancer research have detailed the preparation of solid tumor specimens. After cancer is diagnosed, tumor tissues are removed by biopsy and treated immediately to preserve single cells. Tumor tissue is usually cut into sections of approximately 1 mm3 and washed with PBS to remove fat, visible vessels, and surrounding necrotic areas [6] should be excluded before subsequent bioinformatics analysis to mitigate its influence on downstream analysis results. The percentage of mitochondrial reads is a common quality control metric [15]. When there are a large number of mitochondrial transcripts, it means that the cells are in a state of stress [16], so a threshold is commonly applied to exclude data from cells with too many mitochondrial transcripts. Similarly, the proportion of ribosomal reads is another commonly used quality control metric. Because scRNA-seq is mainly used to study functional (messenger) RNA, cells that have had their ribosomes removed and still have a high proportion of ribosome reads cannot be further analyzed [17]. In addition to the quality control methods for a single dataset, a method denoted ‘scRNABatchQC’ has been proposed that facilitates quality assessment across datasets to intuitively detect biases and outliers [18]. To aid researchers who are intimidated by scRNA-seq analysis, Etherington et al. [19] developed tools and training materials that can be used for scRNA-seq training and quality control.
Batch-effect correction
During the scRNA-seq experimental procedure, when cells subject to different conditions are cultured, captured, and sequenced separately, batch effects will be evident [20]. There are several methods available for batch-effect correction of scRNA-seq data, including Seurat 3 [21], MMD-ResNet [22], Harmony [23], Scanorama [24], Liger [25], scMerge [26], ZINB-WaVE [27], and others. Based on a variety of evaluation indicators, Harmony, Liger, and Seurat 3 are the recommended methods for dealing with batch effects, among which Harmony is the first choice due to its shorter run time [28]. Recently, a novel numerical algorithm for batch-effect correction of bulk and scRNA-seq data was proposed, denoted ‘scBatch’. This approach is not limited by the hypothesis of the batch-effect generation mechanism, and is superior to the benchmark batch-effect correction algorithms [29].
Normalization
Data normalization is essential for scRNA-seq to make gene expression comparable within and/or between samples. A number of methods have been developed for the normalization of RNA-seq data [30,31,32,33]. However, the majority of methods follow the same principle as bulk RNA-seq normalization and, thus, are not applicable to scRNA-seq data [30, 31]. Nevertheless, several methods have recently been devised to normalize scRNA-seq data, such as SCONE [34] and regularized negative binomial regression [35]. SCONE provides a flexible framework for users to choose appropriate normalization methods. Normalization using regularized negative binomial regression effectively eliminates technical differences due to different sequencing depth without inhibiting biological heterogeneity. A previous study [36] compared seven scRNA-seq data normalization methods with regard to reduction of noise or bias, and found that each of these methods was suitable to normalize specific types of data for further downstream analysis.
Cell cycle phase assignment
Determining the cell cycle phase of a single cell can facilitate understanding of biological processes such as tumorigenesis [37,38,39,40,41] and cell differentiation [42, 43], and avoid the confounding effects caused by the cell cycle phase prior to downstream analysis. Scialdone et al. [44] described and compared six supervised cell cycle prediction methods based on a cell transcriptome, of which the parameter-free PCA-based method and the custom predictor known as the “Pairs” method performed best in allocating cells to the correct cell cycle stage. Buettner et al. [45] proposed a calculation method, denoted ‘single-cell latent variable model’ (scLVM), which can be used to eliminate variations caused by cell cycle and other confounding factors before downstream analysis. Recently, Hsiao et al. [46] proposed a new method to characterize the progress of the cell cycle, which is different from the traditional classification of cells according to the standard of cell cycle stage (G1, S or G2/M phase), but can quantify the cell cycle progression of induced pluripotent stem cells on a continuum, which provides a basis for the characterization of the cell cycle in other cell types.
Cell clustering
One of the basic goals of scRNA-seq data analysis is to identify cell types from experimental samples to elucidate tissue complexity and heterogeneity. Due to the importance of cell type recognition, efforts have been made to develop new algorithms, including CountClust [47], CIDR [48], SIMLR [49], SAFE [50], and other advanced methods. A few studies [51,52,53,54] have compared and summarized diverse clustering algorithms for scRNA-seq data analysis. Unlike previous methods, Geddes et al. [55] proposed the first ensemble clustering framework based on autoencoder dimension reduction, which could be combined with different clustering algorithms to promote the accurate recognition of cell types. Since then, several new clustering methods have emerged, including DivBiclust [56], a biclustering-based framework, SAME [57], which extracts cluster solutions from multiple methods, and PARC [58], which is suitable for large-scale single-cell data. To overcome flaws associated with the manual labeling of cell types, Shao et al. [59] developed an automatic annotation toolkit, denoted ‘scCATCH’, based on clustering, which can accurately annotate cell types with acceptable repeatability.
Reconstruction of cell trajectory and pseudo-time
By reconstructing cell trajectory and pseudo-time based on scRNA-seq data, dynamic processes in cells can be calculated and simulated, which is of great significance for understanding the transition between cell states in cancer [60]. Currently available algorithms include Monocle 2 [61], Monocle 3 [62], TSCAN [63], Slingshot [64], SLICE [65], LISA [66], p-Creode [67], Waddington-OT [68] and others. Considering the exponential growth in the size of scRNA-seq data, Chen et al. [66] proposed an unsupervised method, denoted ‘Lisa’, for the reconstruction of cell trajectory and pseudo-time for a large number of scRNA-seq datasets. p-Creode is another unsupervised algorithm that can predict cell state-transition trajectories. Waddington-OT uses the mathematical method of optimal transport (OT) to infer ancestor-descendant fate, and reconstruct cell trajectories. In addition to the three methods mentioned above, the algorithms Monocle 2 (the new version Monocle 3), TSCAN, and Slingshot have been shown to have good performance at reconstructing cell trajectory and presudo-time [69].
Differential expression and gene set enrichment analysis
One of the most common uses of gene expression data is for the identification of differentially expressed (DE) genes under different experimental conditions (e.g., stimulated versus non-stimulated, mutant versus wild-type, or between different time points), and thus to determine the root cause of phenotypic differences observed under different conditions [70]. A zero value for a gene’s expression level in scRNA-seq data may indicate two things. One is the “real” zero, caused by the changing characteristics of single-cell gene transcription, while the other is the “dropout” zero, caused by technical reasons, which often affects the validity of differential expression analysis. Miao et al. [71] developed the R package DESingle, which can accurately distinguish between the two types of zeros. DECENT is a DE gene analysis method based on UMI scRNA-seq data, and is used to analyze the pre-dropout distributions of inferred RNA molecules [72]. In addition to dropout zeros, another challenge in differential expression analysis of scRNA-seq data is multimodal data distribution. ZIAQ is the first approach to consider both dropout rates and the complex distributions of scRNA-seq data, which can be used to identify more DE genes [73].
We usually group DE genes according to their participation in common biological processes to facilitate the interpretation of results [74]. Existing gene set enrichment (GSE) analysis methods include DAVID [75], PAGE [76], CAMERA [77] and others, but almost all of these methods are more suitable for bulk RNA-seq analysis [78]. In addition, almost all existing GSE methods are used as a separate step after DE analysis. Considering the above shortcomings, Ma et al. [79] proposed IDEA, a computational method integrating DE analysis and GSE analysis for scRNA-seq, which could greatly improve the outcomes of both.
Gene regulatory network inference
The combination of active transcription factors and their target genes is usually described in gene regulatory networks (GRNs). Revealing these regulatory interactions is the goal of GRN inference methods, providing valuable insights for the identification of causal regulatory factors in biological processes [74]. A class of GRN inference methods are based on Boolean network models, such as SCNS toolkit [80] and BTR [81]. Another approach for the inference of regulatory networks is based on co-expression analysis, and example models include SINCERA [82], which is specifically used for scRNA-seq data. In addition, there are algorithms based on ordinary differential equations, such as SCODE [83] and InferenceSnapshot [84]. Recently, Moerman et al. [85] proposed the GRNBoost2 and Arboreto frameworks, which can help researchers to deduce high-quality GRNs from large datasets in a reasonable amount of time.
Progress of single-cell RNA sequencing in tumors
Cancer patients may be unresponsive to therapy due to drug resistance and metastasis of single cells, both of which constitute major challenges in the treatment of malignant tumors. About 90% of available drugs are effective in less than half of patients [86]. Cancer is associated with the interaction of thousands of gene products, and genotype as well as interactions vary greatly within and between tumors [87], which is a key reason for the failure of some drugs. In contrast to bulk analysis, which does not account for the differences between cancer cells and their cancer-related counterparts, scRNA-seq provides unprecedented high resolution for the analysis of each individual malignant cell, stromal cell, endothelial cell, parenchymal cell, and immune cell, as well gene expression and pathway activation [88]. Thus, scRNA-seq provides insights that contribute to the development of strategies for cancer treatment and personalized medicine (see Additional file 1).
Highlighting intra- and inter-tumoral heterogeneity
The considerable heterogeneity of tumors and tumor tissue samples between different patients is an important reason for treatment failure [89]. Therefore, understanding the functional status of individual tumor cells and recognizing cell subset composition and characteristics is of great significance for cancer biology and treatment strategies (Table 1 (see Additional file 2)).
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults [90]. It is the glioma with the highest degree of malignancy, and the most often seen in clinical practice, with poor prognosis and a lack of effective treatment regimens [91]. In 2014, Patel et al. [92] analyzed 430 cells of 5 primary GBMs (all IDH1/2 wild-type primary GBMs) using scRNA-seq, and found that these cells differed in the expression of various programs related to carcinogenic signaling pathways, cell proliferation, the immune response, and hypoxic stress. In 2018, Yuan et al. [93] analyzed high-grade glioma (HGG) with large-scale parallel scRNA-seq using a high-density microwell system, and found that, similar to oligodendrocyte progenitors, glioma cells exhibited proliferative characteristics. In contrast, similar to astrocytes, neuroblasts, and oligodendrocytes, glioma cells exhibited an amitotic state in tumors.
Melanoma is a highly malignant skin cancer with four clinically distinguishable subtypes and is responsible for approximately half of skin cancer-related deaths in Japan [94]. Gerber et al. [95] used scRNA-seq to analyze transcription in cells from three different metastatic melanoma patients (BRAF/NRAS wild type, BRAF mutant/NRAS wild type, and BRAF wild type /NRAS mutant). BRAF/NRAS wild-type samples had a low-abundance subgroup with high expression of ABC transporters, while cells from the other two samples exhibited more homogeneous single-cell gene expression patterns.
Head and neck squamous cell carcinoma (HNSCC) encompasses a group of malignant tumors originating from the squamous epithelium of the oral cavity, oropharynx, larynx, and hypopharynx [Discovery of invasion and metastasis mechanisms The ability of single-cell gene expression profiling to identify specific patterns of gene expression allows for the elucidation of mechanisms underlying tumor invasion and metastasis [154]. The combination of scRNA-seq with genoty** can more accurately distinguish malignant cells from normal cells [101]. In the future, different omics technologies could combine with scRNA-seq technology to more comprehensively characterize individual cells. With deepening understanding of the cellular dynamics of cancer, the efficacy of personalized medicine will improve, ultimately saving lives and reducing the global burden of cancer on healthcare systems.
Availability of data and materials
Not applicable.
Abbreviations
- scRNA-seq:
-
Single-cell RNA sequencing
- scLVM:
-
Single-cell latent variable model
- OT:
-
Optimal transport
- DE:
-
Differentially expressed
- GSE:
-
Gene set enrichment
- GRN:
-
Gene regulatory network
- GBM:
-
Glioblastoma
- HGG:
-
High-grade glioma
- LC:
-
Lung cancer
- SCLC:
-
Small-cell lung carcinoma
- NSCLC:
-
Non-small-cell lung carcinoma
- TME:
-
Tumor microenvironment
- AML:
-
Acute myeloid leukemia
- CTC:
-
Circulating tumor cell
- p-EMT:
-
Partial epithelial-to-mesenchymal transition
- TNBC:
-
Triple negative breast cancer
- MM:
-
Multiple myeloma
- cPC:
-
Circulating plasma cell
- EMP:
-
Extramedullary plasmacytoma
- PDX:
-
Patient-derived xenograft
- LUAD:
-
Lung adenocarcinoma
- MB:
-
Medulloblastoma
- SHH:
-
Sonic Hedgehog
- HCC:
-
Hepatocellular carcinoma
- TAM:
-
Tumor-associated macrophage
- CRC:
-
Colorectal cancer
- HCMB:
-
Human cytomegalovirus
- G34R/V:
-
Glycine 34 to arginine or valine
- RCC:
-
Renal cell carcinoma
- MRCC:
-
Metastatic renal cell carcinoma
- MNB:
-
Multilayer network biomarker
- OPC:
-
Oligodendrocyte precursor cell
- NPC:
-
Nasopharyngeal carcinoma
- EGFR:
-
Epidermal growth factor receptor
References
Williams MJ, Werner B, Heide T, et al. Quantification of subclonal selection in cancer from bulk sequencing data. Nat Genet. 2018;50(6):895–903. https://doi.org/10.1038/s41588-018-0128-6.
Tang F, Barbacioru C, Wang Y, et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. https://doi.org/10.1038/nmeth.1315.
Suva ML, Tirosh I. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75(1):7–12. https://doi.org/10.1016/j.molcel.2019.05.003.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–96. https://doi.org/10.1126/science.aad0501.
Shalek AK, Satija R, Shuga J, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510(7505):363–9. https://doi.org/10.1038/nature13437.
Haque A, Engel J, Teichmann SA, Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. https://doi.org/10.1186/s13073-017-0467-4.
Zhao J, Guo C, ** heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget. 2017;8(1):846–62. https://doi.org/10.18632/oncotarget.13666.
Fan C, Wang J, Tang Y, Zhang S, **ong F, Guo C, et al. Upregulation of long non-coding RNA LOC284454 may serve as a new serum diagnostic biomarker for head and neck cancers. BMC Cancer. 2020;20(1):917. https://doi.org/10.1186/s12885-020-07408-w.
Ban Y, Tan P, Cai J, et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(6):1282–96. https://doi.org/10.1002/1878-0261.12676.
Yi M, Tan Y, Wang L, et al. TP63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development. Cell Mol Life Sci. 2020;77(21):4325–46. https://doi.org/10.1007/s00018-020-03539-2.
Deng X, **ong W, Jiang X, Zhang S, Li Z, Zhou Y, et al. LncRNA LINC00472 regulates cell stiffness and inhibits the migration and invasion of lung adenocarcinoma by binding to YBX1. Cell Death Dis. 2020;11(11):945. https://doi.org/10.1038/s41419-020-03147-9.
Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–89. https://doi.org/10.1038/s41591-018-0096-5.
van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176(6):1265–1281 e1224. https://doi.org/10.1016/j.cell.2019.01.031.
Fan C, Qu H, **ong F, Tang Y, Tang T, Zhang L, et al. CircARHGAP12 promotes nasopharyngeal carcinoma migration and invasion via ezrin-mediated cytoskeletal remodeling. Cancer Lett. 2021;496:41–56. https://doi.org/10.1016/j.canlet.2020.09.006.
Tang L, **ong W, Zhang L, et al. circSETD3 regulates MAPRE1 through miR-615-5p and miR-1538 sponges to promote migration and invasion in nasopharyngeal carcinoma. Oncogene. 2021;40(2):307–21. https://doi.org/10.1038/s41388-020-01531-5.
Wu Y, Wang D, Wei F, et al. EBV-miR-BART12 accelerates migration and invasion in EBV-associated cancer cells by targeting tubulin polymerization-promoting protein 1. FASEB J. 2020;34(12):16205–23. https://doi.org/10.1096/fj.202001508r.
Tang T, Yang L, Cao Y, et al. LncRNA AATBC regulates Pinin to promote metastasis in nasopharyngeal carcinoma. Mol Oncol. 2020;14(9):2251–70. https://doi.org/10.1002/1878-0261.12703.
Wu C, Li M, Meng H, Liu Y, Niu W, Zhou Y, et al. Analysis of status and countermeasures of cancer incidence and mortality in China. Sci China Life Sci. 2019;62(5):640–7. https://doi.org/10.1007/s11427-018-9461-5.
Ting David T, Wittner Ben S, Ligorio M, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014;8(6):1905–18. https://doi.org/10.1016/j.celrep.2014.08.029.
Mermer G, Turk M. Assessment of the effects of breast cancer training on women between the ages of 50 and 70 in Kemalpasa, Turkey. Asian Pac J Cancer Prev. 2014;15(24):10749–55. https://doi.org/10.7314/apjcp.2014.15.24.10749.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. https://doi.org/10.1002/ijc.29210.
Chung W, Eum HH, Lee H-O, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8(1):15081. https://doi.org/10.1038/ncomms15081.
Brigle K, Rogers B. Pathobiology and diagnosis of multiple myeloma. Semin Oncol Nurs. 2017;33(3):225–36. https://doi.org/10.1016/j.soncn.2017.05.012.
Geng S, Wang J, Zhang X, et al. Single-cell RNA sequencing reveals chemokine self-feeding of myeloma cells promotes extramedullary metastasis. FEBS Lett. 2019;594(3):452–65. https://doi.org/10.1002/1873-3468.13623.
Lee MC, Lopez-Diaz FJ, Khan SY, et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U S A. 2014;111(44):E4726–35. https://doi.org/10.1073/pnas.1404656111.
Kim KT, Lee HW, Lee HO, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015;16:127. https://doi.org/10.1186/s13059-015-0692-3.
Chen X, Wen Q, Stucky A, et al. Relapse pathway of glioblastoma revealed by single-cell molecular analysis. Carcinogenesis. 2018;39(7):931–6. https://doi.org/10.1093/carcin/bgy052.
Millard NE, De Braganca KC. Medulloblastoma. J Child Neurol. 2016;31(12):1341–53. https://doi.org/10.1177/0883073815600866.
Smoll NR, Drummond KJ. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci. 2012;19(11):1541–4. https://doi.org/10.1016/j.jocn.2012.04.009.
Ocasio J, Babcock B, Malawsky D, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun. 2019;10(1):5829. https://doi.org/10.1038/s41467-019-13657-6.
Wei X, Chen Y, Jiang X, et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol Cancer. 2021;20(1):7. https://doi.org/10.1186/s12943-020-01288-1.
Jiang X, Wang J, Deng X, **ong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Research. 2020;39(1):204. https://doi.org/10.1186/s13046-020-01709-5.
Fan C, Zhang S, Gong Z, et al. Emerging role of metabolic reprogramming in tumor immune evasion and immunotherapy. Sci China Life Sci. 2021;64(4):534-547. https://doi.org/10.1007/s11427-019-1735-4.
Zhu K, Li P, Mo Y, Wang J, Jiang X, Ge J, et al. Neutrophils: accomplices in metastasis. Cancer Lett. 2020;492:11–20. https://doi.org/10.1016/j.canlet.2020.07.028.
Wei F, Wang D, Wei J, et al. Metabolic crosstalk in the tumor microenvironment regulates antitumor immunosuppression and immunotherapy resisitance. Cell Mol Life Sci. 2021;78(1):173–93. https://doi.org/10.1007/s00018-020-03581-0.
Jiang X, Wang J, Deng X, **ong F, Ge J, **ang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):10. https://doi.org/10.1186/s12943-018-0928-4.
Wang YA, Li XL, Mo YZ, Fan CM, Tang L, **ong F, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17(1):168. https://doi.org/10.1186/s12943-018-0913-y.
Duan S, Guo W, Xu Z, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. 2019;18(1):29. https://doi.org/10.1186/s12943-019-0956-8.
Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18(1):128. https://doi.org/10.1186/s12943-019-1055-6.
Ren D, Hua Y, Yu B, et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol Cancer. 2020;19(1):19. https://doi.org/10.1186/s12943-020-1144-6.
Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67(1):103–17. https://doi.org/10.1146/annurev-med-090514-013832.
Muller S, Kohanbash G, Liu SJ, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234. https://doi.org/10.1186/s13059-017-1362-4.
Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750–765 e717. https://doi.org/10.1016/j.cell.2017.04.014.
Azizi E, Carr AJ, Plitas G, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174(5):1293–1308 e1236. https://doi.org/10.1016/j.cell.2018.05.060.
Bo H, Fan L, Li J, et al. High expression of lncRNA AFAP1-AS1 promotes the progression of colon cancer and predicts poor prognosis. J Cancer. 2018;9(24):4677–83. https://doi.org/10.7150/jca.26461.
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268–72. https://doi.org/10.1038/s41586-018-0694-x.
Johnson TS, Abrams ZB, Mo X, Zhang Y, Huang K. Lack of human cytomegalovirus expression in single cells from glioblastoma tumors and cell lines. J Neuro-Oncol. 2017;23(5):671–8. https://doi.org/10.1007/s13365-017-0543-y.
Tirosh I, Venteicher AS, Hebert C, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309–13. https://doi.org/10.1038/nature20123.
Venteicher AS, Tirosh I, Hebert C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355(6332):eaai8478. https://doi.org/10.1126/science.aai8478.
Saurty-Seerunghen MS, Bellenger L, El-Habr EA, et al. Capture at the single cell level of metabolic modules distinguishing aggressive and indolent glioblastoma cells. Acta Neuropathol Commun. 2019;7(1):155. https://doi.org/10.1186/s40478-019-0819-y.
Hovestadt V, Smith KS, Bihannic L, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572(7767):74–9. https://doi.org/10.1038/s41586-019-1434-6.
Weng Q, Wang J, Wang J, et al. Single-cell transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell. 2019;24(5):707–723 e708. https://doi.org/10.1016/j.stem.2019.03.006.
Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, et al. Histone H3.3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell. 2020;183(6):1617–1633 e1622. https://doi.org/10.1016/j.cell.2020.11.012.
Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34(3):193–205. https://doi.org/10.1016/j.ctrv.2007.12.001.
Kim KT, Lee HW, Lee HO, et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016;17:80. https://doi.org/10.1186/s13059-016-0945-9.
Zhang J, Guan M, Wang Q, Zhang J, Zhou T, Sun X. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas. Brief Bioinform. 2019; 21(3):1080-1097. https://doi.org/10.1093/bib/bbz040.
Darmanis S, Sloan SA, Croote D, et al. Single-cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017;21(5):1399–410. https://doi.org/10.1016/j.celrep.2017.10.030.
Filbin MG, Tirosh I, Hovestadt V, Shaw MKL, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360(6386):331–5. https://doi.org/10.1126/science.aao4750.
Wang Q, Tan Y, Fang C, et al. Single-cell RNA-seq reveals RAD51AP1 as a potent mediator of EGFRvIII in human glioblastomas. Aging. 2019;11(18):7707–22. https://doi.org/10.18632/aging.102282.
Cai J, Chen S, Yi M, et al. ΔNp63α is a super enhancer-enriched master factor controlling the basal-to-luminal differentiation transcriptional program and gene regulatory networks in nasopharyngeal carcinoma. Carcinogenesis. 2020;41(9):1282–93. https://doi.org/10.1093/carcin/bgz203.
Chen S, Youhong T, Tan Y, et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR2. Carcinogenesis. 2020;41(6):723–33. https://doi.org/10.1093/carcin/bgz180.
Fan C, Tu C, Qi P, et al. GPC6 promotes cell proliferation, migration, and invasion in nasopharyngeal carcinoma. J Cancer. 2019;10(17):3926–32. https://doi.org/10.7150/jca.31345.
Mo Y, Wang Y, **ong F, Ge X, Li Z, Li X, et al. Proteomic analysis of the molecular mechanism of lovastatin inhibiting the growth of nasopharyngeal carcinoma cells. J Cancer. 2019;10(10):2342–9. https://doi.org/10.7150/jca.30454.
**ong F, Deng S, Huang HB, et al. Effects and mechanisms of innate immune molecules on inhibiting nasopharyngeal carcinoma. Chin Med J. 2019;132(6):749–52. https://doi.org/10.1097/cm9.0000000000000132.
Zhang Y, Wang D, Peng M, et al. Single-cell RNA sequencing in cancer research. J Exp Clin Cancer Res. 2021;40(1):81. https://doi.org/10.1186/s13046-021-01874-1.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8. https://doi.org/10.1038/nmeth.4380.
Acknowledgements
Not applicable.
Funding
This work was supported partially by grants from the National Natural Science Foundation of China (81903138, 81972776, 81803025, 81772928, 81702907, 81772901, 81672993, 81672683), and the Natural Science Foundation of Hunan Province (2019JJ50778, 2018SK21210, 2018SK21211, 2018JJ3704, 2018JJ3815).
Author information
Authors and Affiliations
Contributions
LLY, XF, WYM, ZSS, GZJ, LXY, HY, SL, WFY, LQJ, XB, ZM, LXL collected the related paper and drafted the manuscript. LY, LGY, ZZY, XW, GC participated in the design of the review and draft the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Fig. 1. Application of scRNA-seq in tumor research. ScRNA-seq can be used to analyze the proportion of various immune cell types and immune repertoire characteristics, guiding choices related to immunotherapy. The analysis of single tumor cells can improve our understanding of drug resistance, invasion, and metastasis mechanisms, as well as cell origin and evolution, in turn aiding the choice of targeted drugs. T, T cell; B, B cell; NK, natural killer cell; TAM, tumor-associated macrophage; Th, helper T cell; Tc, cytotoxic T cell; Treg, regulatory T cell; Tm, memory T cell; Ts, suppressor T cell; B1, T cell independent B cell; B2, T cell dependent B cell.
Additional file 2
: Table 1. Overview of related studies using scRNA-seq.
Additional file 3
: Table 2. Overview of related articles using scRNA-seq.
Additional file 4
: Table 3. Overview of related studies using scRNA-seq.
Additional file 5
: Table 4. Overview of related articles using scRNA-seq.
Additional file 6
: Table 5. Overview of related articles using scRNA-seq.
Additional file 7
: Table 6. Overview of related articles using scRNA-seq.
Additional file 8
: Table 7. Overview of related articles using scRNA-seq.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, L., **ong, F., Wang, Y. et al. What are the applications of single-cell RNA sequencing in cancer research: a systematic review. J Exp Clin Cancer Res 40, 163 (2021). https://doi.org/10.1186/s13046-021-01955-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-01955-1