
Abstract
Background
Emerging evidence has shown long noncoding RNAs (lncRNAs) exert important roles in colorectal cancer (CRC) tumorigenesis. However, most lncRNAs involved in this process remain undefined and the underlying molecular mechanisms mediated by lncRNAs are largely unknown.
Methods
An unbiased screening was used to identify novel lncRNAs involved in CRC according to an online-available data dataset. In situ hybridization (ISH) and qRT-PCR was used to detect lncRNA expression patterns. CCK8, colony formation, fluorescence activated cell sorter (FACS), transwell, xenograft nude mouse model and western blot assays were used to analyze the functions of SLCO4A1-AS1. RNA-pulldown, western blot, RNA fluorescence in situ hybridization (RNA-FISH) and electrophoretic mobility shift assay (EMSA) assays were utilized to explore the molecular mechanism of SLCO4A1-AS1.
Results
LncRNA SLCO4A1-AS1 was significantly upregulated in CRC tissues and its overexpression was closely related with poor prognosis and tumor metastasis. By knocking down SLCO4A1-AS1, we found that SLCO4A1-AS1 promoted the proliferation, migration, invasion and epithelial–mesenchymal transition (EMT) of CRC cells in vitro, as well as inhibited cell apoptosis. Moreover, SLCO4A1-AS1 dramatically delayed tumor propagation in vivo. Mechanistically, SLCO4A1-AS1 activates Wnt/β-catenin signaling. SLCO4A1-AS1 enhanced the stability of β-catenin by impairing the interaction of β-catenin with GSKβ and inhibiting its phosphorylation. Finally, restoration of β-catenin protein level rescued the proliferation, migration and invasion in SLCO4A1-AS1-depleted CRC cells.
Conclusion
SLCO4A1-AS1 serves as an oncogenic role in CRC through activating Wnt/β-catenin signaling pathway. And SLCO4A1-AS1 might be a useful biomarker for CRC diagnosis and prognosis.
Similar content being viewed by others
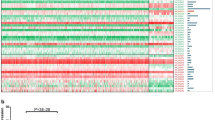


Background
As one of the most prevalent cancers, colorectal cancer (CRC) has become the third-leading cause of cancer-related death worldwide every year [1]. Hyperactivation of some signaling pathways, including wnt/β-catenin, PI3K/AKT, JAK/STAT signaling pathways and so on, often contributes to the development, progression, metastasis and resistance to chemotherapy of CRC [5,6,7]. Emerging evidence shows that lncRNAs are critical regulators involved in various biological processes via multiple mechanisms [8, 9], such as development, immune regulation and especially tumorigenesis [10,11,12]. Importantly, accumulating studies have proven that aberrant expression of lncRNAs is closely related to various human cancers [4f). Moreover, deletion of this region (nt 900~ 1200) abrogated this interaction between SLCO4A1-AS1 and β-catenin (Fig. 4g). Furthermore, we performed RNA electrophoretic mobility shift assay (RNA-EMSA) with biotin-labeled probe (nt 900~ 1200) and demonstrated their direct association (Fig. 4h).

SLCO4A1-AS1 interacts with β-catenin. a RNA pulldown using biotin-labeled probe or intron control and sample lysates, followed by SDS-PAGE electrophoresis, silver staining and MS identification. b RNA pulldown showed that SLCO4A1-AS1 interacted with β-catenin in HCT116 and SW480 cells. SLCO4A1-AS1 was labeled with biotin. c RNA IP showed that β-catenin enriched SLCO4A1-AS1 in HCT116 and SW480 cell lysates. d β-catenin enriched SLCO4A1-AS1 in CRC sample cell lysates. e SLCO4A1-AS1 co-localized with β-catenin in CRC sample cells as shown by RNA fluorescence in situ hybridization (RNA-FISH). The scale bar was 10 μm. f, g Domain map** showed that SLCO4A1-AS1 (nt: 900~ 1200) interacted with β-catenin and was indispensible. d900~ 1200 represented deletion of nt 900~ 1200. h RNA electrophoretic mobility shift assay (RNA-EMSA) showed that biotin-labeled SLCO4A1-AS1 (nt: 900~ 1200) directly bond to β-catenin. *p < 0.05 and **p < 0.01
SLCO4A1-AS1 increased the stability of β-catenin by inhibiting its phosphorylation
We have confirmed the interaction between SLCO4A1-AS1 and β-catenin. Then we performed western blot and found that SLCO4A1-AS1 knockdown significantly decreased the protein level of β-catenin in HCT116 and SW480 cells (Fig. 5a). On the contrary, overexpression of full-lengthen or nt 900~ 1200 dramatically upregulated the protein level of β-catenin in HCT116 and SW480 cells (Fig. 5b). Additionally, we validated the elevated β-catenin ubiquitination signals using β-catenin immunoprecipitates from SLCO4A1-AS1–depleted HCT116 cells through (Fig. 5c) and consequently decreased β-catenin stability (Fig. 5d). A previous study showed that β-catenin phosphorylation by GSKβ promotes its ubiquitination-mediated degradation [30]. We then assessed the effect of SLCO4A1-AS1 on β-catenin phosphorylation and found that SLCO4A1-AS1 knockdown significantly increased β-catenin phosphorylation in HCT116 and SW480 cells (Fig. 5e). Moreover, SLCO4A1-AS1-overexpressed CRC sample tissues showed lower β-catenin phosphorylation (Fig. 5f). Besides, we found that SLCO4A1-AS1 knockdown enhanced the interaction between β-catenin and GSKβ in HCT116 and SW480 cells (Fig. 5g) while overexpressing SLCO4A1-AS1 abrogated their interaction (Fig. 5h). To further determine whether SLCO4A1-AS1 activated Wnt/β-catenin signaling by enhancing the stability of β-catenin, we restored the protein levels of β-catenin in HCT116 and SW480 cells (Fig. 5i). By qRT-PCR, we found that restoration of β-catenin rescued the activation of Wnt/β-catenin signaling in HCT116 and SW480 cells (Fig. 5j). Summarily, our results indicated that SLCO4A1-AS1 stabilized β-catenin by preventing the association between β-catenin and GSKβ, and consequently activated Wnt/β-catenin signaling in CRC.

SLCO4A1-AS1 increased the stability of β-catenin by inhibiting its phosphorylation. a WB analysis showed that siRNA-induced SLCO4A1-AS1 knockdown decreased the protein level of β-catenin in HCT116 and SW480 cells. H3, nuclear marker; EEA1, cytoplasmic marker. b Overexpression of SLCO4A1-AS1 (full-length or nt 900~ 1200) promoted the protein level of β-catenin in HCT116 and SW480 cells. H3, nuclear marker; EEA1, cytoplasmic marker. c Knockdown of SLCO4A1-AS1 enhanced the ubiquitination of β-catenin in HCT116 cells. d SLCO4A1-AS1 knockdown accelerated the degradation of β-catenin in HCT116 and SW480 cells. Chx, cycloheximide. e SLCO4A1-AS1 knockdown promoted the phosphorylation of β-catenin in HCT116 and SW480 cells as shown by western blotting. f WB analysis showed that the protein levels of β-catenin were higher in SLCO4A1-AS1high CRC samples while the phosphorylation of β-catenin was lower. g SLCO4A1-AS1 knockdown enhanced the interaction between β-catenin and GSK3β in HCT116 and SW480 cells. h WB analysis indicated that overexpression of SLCO4A1-AS1 (full-length or nt 900~ 1200) abrogated the interaction between β-catenin and GSK3β in HCT116 and SW480 cells. i, j Restoration of the protein levels of β-catenin by ectopic expression of β-catenin (i) rescued SLCO4A1-AS1 knockdown-induced inactivation of wnt/β-catenin signaling in HCT116 and SW480 cells (j)
SLCO4A1-AS1 promotes CRC proliferation, migration and invasion by activating wnt/β-catenin signaling in vitro and in vivo
Whether the SLCO4A1-AS1-mediated augment of CRC cell growth and metastasis relied on activation of Wnt/β-catenin signaling was assessed in SLCO4A1-AS1-silenced HCT116 and SW480 cells transfected with β-catenin-overexpressing plasmid or empty control. Results showed that decreased proliferation, colony formation, migration and invasion potentials of SLCO4A1-AS1-silenced cells were rescued by ectopic expression of β-catenin in HCT116 and SW480 cells (Fig. 6a-d). What’s more, SLCO4A1-AS1 knockdown delayed tumor growth in vivo while overexpression of β-catenin in the meantime reversed it (Fig. 6e and f). Then we measured the activation of Wnt/β-catenin signaling in formed tumor tissues. As shown, the Wnt/β-catenin signaling was also downregulated in vivo after SLCO4A1-AS1 depletion (Fig. 6g). Finally, we evaluated the effect of SLCO4A1-AS1 on tumor metastasis in vivo, and found that SLCO4A1-AS1 knockdown severely reduced the metastatic nodules in the liver while β-catenin overexpression reversed this trend (Fig. 6h and i). Taken together, above data suggested that SLCO4A1-AS1 exerted functions dependent on activation of Wnt/β-catenin signaling in CRC.

SLCO4A1-AS1 promotes CRC proliferation, migration and invasion by activating wnt/β-catenin signaling in vitro and in vivo. a, b Ectopic expression of β-catenin rescued the decreased proliferation ability of HCT116 and SW480 cells induced by SLCO4A1-AS1 knockdown as shown by CCK-8 and colony formation assays. c, d Overexpression of β-catenin restored the migration and invasion potential of HCT116 and SW480 cells as indicated by a Transwell and Matrigel assay, respectively. Scale bar, 50 μm. e, f SLCO4A1-AS1 knockdown delayed the tumor propagation in vivo while overexpressing β-catenin rescued it. Tumor volumes were measured at indicative time points. Tumor weights were measured at the end point of experiments. g Total RNAs were extracted from tumor tissues in f and the activation of wnt/β-catenin signaling was assessed by qRT-PCR. Results showed that SLCO4A1-AS1 knockdown downregulated wnt/β-catenin signaling while ectopic expression of β-catenin upregulated it in vivo. h Knockdown of SLCO4A1-AS1 expression remarkably reduced the metastatic nodules in the liver. i The liver sections were shown via H&E staining. Scale bar, 50 μm. *p < 0.05 and **p < 0.01
Discussion
In recent years, great efforts have been made to search cancer-related lncRNAs and determine their molecular mechanisms in tumor development and progression [31]. Here we identified the physiological functions of an uncharacterized lncRNA SLCO4A1-AS1 and determined its molecular mechanism. SLCO4A1-AS1 was highly expressed in CRC tissues and may act as a biomarker for CRC diagnosis. Notably, we detected an unbelievable high AUC, which might be due to the limited size of CRC samples. SLCO4A1-AS1 was found to promote the proliferation and invasion of CRC cells, indicating that it may be implicated in the process of tumorigenesis. Moreover, SLCO4A1-AS1 knockdown induced CRC cell apoptosis, which implied that SLCO4A1-AS1 may be important for the functional maintenance of normal cancer cells.
Further analysis showed that the activation of Wnt/β-catenin signaling was affected by SLCO4A1-AS1. As one of the most essential intracellular signaling pathways, Wnt/β-catenin signaling mediates diverse cellular processes, including embryonic development, cell proliferation, differentiation, migration, survival and so on [32,33,34]. Hyperactivation of the Wnt/β-catenin signaling often leads to various cancers such as liver cancer and CRC [35,36,37]. For instance, CRCAT-1-mediated activation of Wnt signaling pathway promotes cell proliferation and inhibits apoptosis in cervical cancer cells [38]. Additionally, activation of Wnt/β-catenin signaling by TGFβ promotes CRC development [39]. In our study, we found that SLCO4A1-AS1 knockdown severely decreased the protein level of β-catenin but not mRNA level by the mechanism that SLCO4A1-AS1 inhibited the phosphorylation and consequently ubiquitylation-mediated degradation of β-catenin. Through interacting with β-catenin, SLCO4A1-AS1 impaired the binding of GSK3β to β-catenin and inhibited β-catenin phosphorylation by GSK3β. Emerging evidence shows that lncRNAs can exert functions by regulation in trans [40]. lncRNAs may associate with proteins to regulate their stability, activity or other properties [11, 41, 42]. Based on above evidence, we proposed that SLCO4A1-AS1 may bind to β-catenin and then shield the interactive domain of β-catenin with GSK3β.
β-catenin level plays a pivot role in the canonical Wnt pathway [43]. Increase of β-catenin protein level may lead to abnormal cell proliferation and human diseases [44]. The regulation of β-catenin protein level is complicated and delicate. Phosphorylation and ubiquitylation of β-catenin are all reported to participate in the regulation of β-catenin stability [45]. For example, Liu et al. demonstrated that phosphorylation of β-catenin by CKIα in vivo is indispensible for subsequent phosphorylation of β-catenin by GSK3β, which finally leads to degradation of β-catenin [45]. Besides, other studies showed that phosphorylated β-catenin is ubiquitylated by E3 ubiquitin ligase β-TrCP and then degraded by the ubiquitin–proteasome pathway [46, 47]. Abrogation of β-catenin degradation promotes the accumulation of β-catenin in cells and induces tumor occurrence. For instance, inactivating mutation of APC, a pivot subunit of the degradation complex of β-catenin, gave rise to spontaneous CRC in mice [48]. So far, the regulatory mechanism of β-catenin turnover is not fully understood. Our study revealed that SLCO4A1-AS1 regulated the stability of β-catenin by weakening the association between β-catenin and GSK3β.
Continuous mutations of genes are popularly considered as a cause of tumors [49]. Gene copy number alterations or mutations are the common aberrances in cancers, and some studies have demonstrated the relevance between gene copy-number alterations and tumor formation and progression [50]. Previous study shows that DNA copy-number gain was observed on chromosome 20q in primary colorectal tumor [51]. Notably, SLCO4A1-AS1 is also located on chromosome 20q. Moreover, SLCO4A1-AS1 is really substantially amplified in CRC according to TCGA database and our experiment (Fig. 1b and c). However, how copy-number amplifications on chromosome 20q affect the expression and functions of SLCO4A1-AS1 in CRC remains further investigation.
Conclusion
In summary, we found that lncRNA SLCO4A1-AS1 was highly expressed in CRC tissues. Upregulated SLCO4A1-AS1 promoted CRC progression through inhibiting the degradation of β-catenin by attenuating the interaction between β-catenin and GSK3β. This study revealed the vital significance of SLCO4A1-AS1 in CRC development.
Abbreviations
- CRC:
-
colorectal cancer
- EMSA:
-
Electrophoretic mobility shift assay
- lncRNA:
-
long noncoding RNA
- RNA-FISH:
-
RNA fluorescence in situ hybridization
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29.
Li T, Lai Q, Wang S, Cai J, **ao Z, Deng D, He L, Jiao H, Ye Y, Liang L, et al. MicroRNA-224 sustains Wnt/beta-catenin signaling and promotes aggressive phenotype of colorectal cancer. J Exp Clin Cancer Res. 2016;35:21.
Song GH, Xu SF, Zhang H, Wang YP, **ao C, Jiang T, Wu LL, Zhang T, Sun X, Zhong L, et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J Exp Clin Cancer Res. 2016;35:148
Wei R, Yang Q, Han B, Li Y, Yao K, Yang X, Chen Z, Yang S, Zhou J, Li M, et al. microRNA-375 inhibits colorectal cancer cells proliferation by downregulating JAK2/STAT3 and MAP3K8/ERK signaling pathways. Oncotarget. 2017;8:16633–41.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8.
Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–10.
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–83.
Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC, Young RA. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci U S A. 2013;110:2876–81.
Karlic R, Ganesh S, Franke V, Svobodova E, Urbanova J, Suzuki Y, Aoki F, Vlahovicek K, Svoboda P. Long non-coding RNA exchange during the oocyte-to-embryo transition in mice. DNA Res. 2017;24:129–41.
Liu B, Ye B, Yang L, Zhu X, Huang G, Zhu P, Du Y, Wu J, Qin X, Chen R, et al. Long noncoding RNA lncKdm2b is required for ILC3 maintenance by initiation of Zfp292 expression. Nat Immunol. 2017;18:499–508.
Liu J, Liu L, Wan JX, Song Y. Long noncoding RNA SNHG20 promotes gastric cancer progression by inhibiting p21 expression and regulating the GSK-3beta/ beta-catenin signaling pathway. Oncotarget. 2017;8:80700–8.
Xu YC, Liang CJ, Zhang DX, Li GQ, Gao X, Fu JZ, **a F, Ji JJ, Zhang LJ, Li GM, Wu JX. LncSHRG promotes hepatocellular carcinoma progression by activating HES6. Oncotarget. 2017;8:70630–41.
Zhu P, Wang Y, Wu J, Huang G, Liu B, Ye B, Du Y, Gao G, Tian Y, He L, Fan Z. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat Commun. 2016;7:13608.
Xu Y, Leng K, Li Z, Zhang F, Zhong X, Kang P, Jiang X, Cui Y. The prognostic potential and carcinogenesis of long non-coding RNA TUG1 in human cholangiocarcinoma. Oncotarget. 2017;8:65823–35.
Zhu J, Shi H, Liu H, Wang X, Li F. Long non-coding RNA TUG1 promotes cervical cancer progression by regulating the miR-138-5p-SIRT1 axis. Oncotarget. 2017;8:65253–64.
Ma ZH, Huang HSY, Wang JR, Zhou Y, Pu FX, Zhao QH, Peng P, Hui BQ, Ji H, Wang KM. Long non-coding RNA SNHG15 inhibits P15 and KLF2 expression to promote pancreatic cancer proliferation through EZH2-mediated H3K27me3. Oncotarget. 2017;8:84153–67.
Yang J, Li C, Mudd A, Gu X. LncRNA PVT1 predicts prognosis and regulates tumor growth in prostate cancer. Biosci Biotechnol Biochem. 2017;81(12):2301–6.
Wang S, Liang K, Hu Q, Li P, Song J, Yang Y, Yao J, Mangala LS, Li C, Yang W, et al. JAK2-binding long noncoding RNA promotes breast cancer brain metastasis. J Clin Invest. 2017;127(12):4498–515
Li J, Xue W, Lv J, Han P, Liu Y, Cui B. Identification of potential long non-coding RNA biomarkers associated with the progression of colon cancer. Oncotarget. 2017;8:75834–43.
Yu XF, Mi L, Dong J, Zou J. Long intergenic non-protein-coding RNA 1567 (LINC01567) acts as a "sponge" against microRNA-93 in regulating the proliferation and tumorigenesis of human colon cancer stem cells. BMC Cancer. 2017;17:716
Huang JZ, Chen M, Chen D, Gao XC, Zhu S, Huang HY, Hu M, Zhu HF, Yan GR. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses Colon Cancer growth. Mol Cell. 2017;68:171.
Shen XG, Bai YF, Luo B, Zhou XG. Upregulation of lncRNA BANCR associated with the lymph node metastasis and poor prognosis in colorectal cancer. Biol Res. 2017;50:32
Liu L, Zhang Y, Wong CC, Zhang J, Dong Y, Li X, Kang W, Chan FKL, Sung JJY, Yu J. RNF6 promotes colorectal Cancer by activating the Wnt/beta-catenin pathway via ubiquitination of TLE3. Cancer Res. 2018;78:1958–71.
Liu BY, Ye BQ, Zhu XX, Huang GL, Yang LL, Zhu PP, Du Y, Wu JY, Meng S, Tian Y, Fan ZS. IL-7R alpha glutamylation and activation of transcription factor Sall3 promote group 3 ILC development. Nat Commun. 2017;8:231
Zhu P, Wang Y, Huang G, Ye B, Liu B, Wu J, Du Y, He L, Fan Z. Lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23:631–9.
Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–6.
Yang Q, Wang Y, Pan X, Ye J, Gan S, Qu F, Chen L, Chu C, Gao Y, Cui X. Frizzled 8 promotes the cell proliferation and metastasis of renal cell carcinoma. Oncotarget. 2017;8:78989–9002.
Bayin NS, Frenster JD, Sen R, Si S, Modrek AS, Galifianakis N, Dolgalev I, Ortenzi V, Illa-Bochaca I, Khahera A, et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget. 2017;8:64932–53.
Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–56.
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–61.
Majidinia M, Aghazadeh J, Jahanban-Esfahlani R, Yousefi B. The roles of Wnt/beta-catenin pathway in tissue development and regenerative medicine. J Cell Physiol. 2018;233(8):5598–612.
De Boer J, Wang HJ, Van Blitterswijk C. Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 2004;10:393–401.
Yang J, Wei D, Wang W, Shen B, Xu S, Cao Y. TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt-beta-catenin signaling pathway. Int J Clin Exp Pathol. 2015;8:11837–46.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80.
White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–32.
Wong CM, Fan ST. Ng IO: beta-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136–45.
Zhang J, Gao YL. CCAT-1 promotes proliferation and inhibits apoptosis of cervical cancer cells via the Wnt signaling pathway. Oncotarget. 2017;8:68059–70.
Wang JL, Qi Z, Li YH, Zhao HM, Chen YG, Fu W. TGF beta induced factor homeobox 1 promotes colorectal cancer development through activating Wnt/beta-catenin signaling. Oncotarget. 2017;8:70214–25.
Rapicavoli NA, Poth EM, Zhu H, Blackshaw S. The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev. 2011;6:32.
Yoon JH, Abdelmohsen K, Kim J, Yang X, Martindale JL, Tominaga-Yamanaka K, White EJ, Orjalo AV, Rinn JL, Kreft SG, et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat Commun. 2013;4:2939.
Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ, Zhang J, Huang H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget. 2015;6:41045–55.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:689–99.
Liu CM, Li YM, Semenov M, Han C, Baeg GH, Tan Y, Zhang ZH, Lin XH, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47.
Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9:207–10.
Aberle H, Bauer A, Stappert J, Kispert A. Kemler R: beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804.
Fodde R, Edelmann W, Yang K, Vanleeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, Kucherlapati R. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–73.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58.
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8.
Nakao K, Mehta KR, Fridlyand J, Moore DH, Jain AN, Lafuente A, Wiencke JW, Terdiman JP, Waldman FM. High-resolution analysis of DNA copy number alterations in colorectal cancer by array-based comparative genomic hybridization. Carcinogenesis. 2004;25:1345–57.
Acknowledgements
The authors thank all patients involved in this study.
Funding
This work was supported by grants from the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2016193) and Harbin medical university scientific research innovation fund (2017LCZX05) and Fund of scientific research innovation of the First Affiliated Hospital of Harbin Medical University (NO.2018B012).
Availability of data and materials
All data and materials can be provided upon request.
Author information
Authors and Affiliations
Contributions
JY performed experiments, analyzed data and wrote the paper; ZHZS, YW and MZ performed some experiments and analyzed data; CS initiated the study, designed experiments and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University. All written informed consents were received from all patients.
Consent for publication
The authors agree for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, J., Han, Z., Sun, Z. et al. LncRNA SLCO4A1-AS1 facilitates growth and metastasis of colorectal cancer through β-catenin-dependent Wnt pathway. J Exp Clin Cancer Res 37, 222 (2018). https://doi.org/10.1186/s13046-018-0896-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-018-0896-y