
Abstract
The current view of neuroplasticity depicts the changes in the strength and number of synaptic connections as the main physical substrate for behavioral adaptation to new experiences in a changing environment. Although transcriptional regulation is known to play a role in these synaptic changes, the specific contribution of activity-induced changes to both the structure of the nucleus and the organization of the genome remains insufficiently characterized. Increasing evidence indicates that plasticity-related genes may work in coordination and share architectural and transcriptional machinery within discrete genomic foci. Here we review the molecular and cellular mechanisms through which neuronal nuclei structurally adapt to stimuli and discuss how the perturbation of these mechanisms can trigger behavioral malfunction.
Similar content being viewed by others

Introduction
In the search for mechanisms that underlie behavioral plasticity, functional and structural changes at synapses are at the core of the theoretical framework. Processes such as long-term potentiation (LTP) or synaptogenesis are thought to be crucial for the adaptation of neuronal circuits to changing environmental conditions [1]. Both stimulus-driven transcriptional responses [2] and different forms of epigenetic regulation [3] are known to participate in these processes. However, only recently high-order chromatin architecture has been implicated in the neurobiology of behavior [4]. Cell biology studies have revealed that the compartmentalization of chromatin dictates the location of specific genes within the neuronal nucleus, thereby conditioning the mechanisms controlling their transcription [5]. The complexity and cellular heterogeneity of neuronal tissue make technically difficult the investigation of the contribution of activity-induced changes in chromatin architecture to neuronal plasticity. However, as technological advances enable deeper insight into the genomic landscape of neurons, increasing evidence indicates that individual genes do not work in isolation; instead, they share niches and machinery within the cell nucleus that sustain coordinated regulation. The levels of regulation include changes in nuclear geometry and subnuclear structures, dynamic interactions of structural proteins and the transcription machinery with chromatin, the relocation of genes into transcriptionally active or repressive areas, and chromatin loo**s that activate regulatory sequences. In the following sections, we review recent studies that have begun to unveil the contribution of these novel mechanisms to neuronal plasticity, and highlight how their malfunction can contribute to the on-set or further development of neuropsychiatric disorders.
Neuronal nuclear structure and its regulation by neuronal activity
In eukaryotic nuclei, DNA is wrapped around an octameric histone core comprising of two copies of each of the canonical histones H2A, H2B, H3 and H4. This basic structure, known as a nucleosome, is repeated along the double-stranded DNA, with a fifth type of histone (the linker histone H1) bridging together consecutive nucleosomes. In this fashion, long DNA strands condense with architectural proteins to form chromatin. Based on the level of compaction we can distinguish three main forms of chromatin. These forms differ biochemically with respect to the presence of specific post-translational modifications (PTMs) at the histone tails and to the binding of structural proteins. Euchromatin, a transcriptionally active form is characterized by permissive marks such as the trimethylation of histone 3 at lysine 4 (H3K4me3), and the acetylation of different lysine residues at the histone tails. In contrast, heterochromatin is a transcriptionally silent form, and is decorated by repressive epigenetic marks. It can be found in two different functional states: constitutive heterochromatin that is characterized by DNA methylation at CpGs and histone H3 trimethylation at lysine 9 (H3K9me3), and facultative heterochromatin, which, as suggested by its name, can harbor transcriptional activity and is marked by H3K27me3 [6].
Although the folding of chromatin fibers during cell division is very similar among all cells [7], the spatial organization of the chromatin in the interphasic nucleus can greatly differ. Thus, during neuronal maturation, centromeric constitutive heterochromatin foci from different chromosomes are reduced in number, and cluster in larger foci known as chromocenters [8, 9] (Fig. 1a). These structures are depleted of the facultative heterochromatin marks H3K27me3 and H3K9me2, and the active isoforms of RNA Polymerase II (RNAPII), indicating that they lack the potential to be transcriptionally active [10]. In parallel to chromocenter formation, chromosome territories are distributed in the interior of the nucleus, defining regions with different gene densities in which gene-poor regions are generally located at the periphery while gene-rich regions are found in the interior of the cell nucleus [11]. Recent studies on the nuclear architecture of chicken neurons have revealed a more extreme form of radial nuclear organization in which chromocenters are radially aligned between the peripheral heterochromatin and DNA-depleted areas in the central nucleoplasm [10]. Notably, some highly specialized neurons, such as the retinal rods of nocturnal mammals, present an inverted distribution of the heterochromatin that could contribute to maximize light transmission trough photoreceptors thereby serving a unique function in nocturnal vision [12].

Nuclear structure and sub-compartments. a. Developmental changes as seen with DAPI staining (in yellow). The nucleus of an embryonic stem cell is euchromatic and relatively homogeneous. Heterochromatin foci (centromeres and telomeres) become more evident in neuronal progenitors. Mature neurons present fewer and denser chromocenters (adapted from microscopy images in [8]). b. Different types of nuclear bodies can be found in the nucleus of post-mitotic neurons
Apart from chromocenters and peripheral heterochromatin, the interphasic neuronal nucleus is structurally complex [13] (Fig. 1b). Based on conventional microscopy techniques, we can define three major components: the nuclear lamina and associated heterochromatin, the nucleoplasm that is defined by a fine and relatively homogeneous granular matrix, and the different internal macrostructures that disrupt this granular matrix. In the following sections we will discuss each of these components and their responses to neuronal activation.
Nuclear envelope and lamina
Nuclear architecture and genome organization depend on the integrity of the nuclear envelope, a boundary that separates the cytoplasm from the nucleoplasmic reticulum. This boundary is composed of two phospholipid bilayers spanned at intervals by proteins that act as nuclear pores. The nuclear envelope is not an inert barrier, it participates in different processes including gene regulation and the transport of ions and macromolecular cargos [14]. Its geometry in neurons is rather plastic and responds to neuronal activity [15]. In the case of hippocampal neurons, there are both spherical and highly infolded nuclei featuring different degrees of complexity, with nuclear infoldings being antagonistically regulated by synaptic and extrasynaptic NMDA receptors [16]. Infolded nuclei typically have larger surfaces accompanied by an increase in nuclear pore complexes (NPC) that facilitates calcium influx and the transport between the nuclear and cytosolic plasmas.
Internally attached to the nuclear envelope is the nuclear lamina, whose main components are the lamin proteins A/C, B1 and B2 [17]. These proteins form a scaffold and bind to peripheral chromatin, playing an essential role in transcriptional regulation. Cellular biology studies have shown that the lamin composition of the nuclear envelope changes throughout neuronal differentiation. While primary progenitors have lamin A/C, B1 and B2 in equal amounts, neuroblasts have more B1 and some B2, and mature neurons preferentially express B2, some A/C, and little B1 [18]. Genetic experiments in mice have demonstrated that lamins B1 and B2, despite their great sequence homology, have unique roles in the develo** brain, and that increased production of one does not compensate for the loss of the other [19, 20].
Lamin-associated chromatin domains (LADs) are enriched in transcriptional and epigenetic repressors [21]. Although the attachment of chromatin to the nuclear lamina has been found to promote transcriptional repression [17], this relationship is not strict. In fact, genes in both the margin and the center can be expressed, although peripheral genes are less likely to be transcribed than inactive genes dissociated from the lamina [51]. The proteins CTCF (aka CCCTC-binding factor), mediator and cohesin are important components of the insulator complex that appear in distinct combinations depending on the range of interaction. CTCF and cohesin locate together in active regulatory sequences where they mediate long-range constitutive interactions. They are fundamental building blocks behind insulated chromosomal neighborhoods containing super-enhancers necessary for cell identity [52]. For instance, the presence of CTCF/cohesin marks megabase-sized TADs whose boundaries are usually constant among all cell types, although there can be cell-type specific subTAD organization [53]. Whereas cohesin is involved in regulation of tissue-specific transcription [54], CTCF plays a prominent role enabling chromatin loo** through the pairing of sequences that contain its binding site [53, 55]. In turn, mediator and cohesin are found in short-range complexes that bridge enhancers and promoters. While mediator is necessary for the loading of enhancers with TFs and the formation of the initiation complex at the promoter [53], cohesin together with the “loader” protein Nipped-B-like protein (NIPBL) and other factors, brings DNA sequences together forming a ring structure that physically promotes their approximation [56]. The involvement of these proteins in neurodevelopment and cognition is supported by the finding that mutations in the encoding genes cause intellectual disability and severe neurodevelopmental defects (see below). Moreover, experiments in mice indicate that CTCF loss throughout developmental stages has been shown to cause neuronal death and deregulate neuronal differentiation [57], while ablation in postmitotic neurons caused growth retardation, abnormal hind-paw clas**, defects in somatosensory cortical maps, and reduced dendritic arborization and spine density [58].
Poised RNAPII and transcription factories
The term transcription factory refers to discrete foci in the eukaryotic nucleus where transcription occurs [59]. These mega-structures promote physical interactions between genes that share the same regulatory machinery, which may enable their synchronous expression [60]. Consistent with this notion, genomic analyses indicate that TF binding can occur in nucleosome-depleted stretches of DNA lacking their canonical binding motifs through the interaction with other TFs and cofactors. Enhancer elements are also thought to form part of these mega transcription factor complexes [61] that are enriched in cohesin binding and strongly labeled with RNAPII antibodies [62]. It has been described in different immortalized human cell lines that loci highly enriched in RNAPII are often associated with looped chromatin in promoter-promoter interactions (the most common) or in the interactions between promoters and distal regulatory elements [61]. Single-gene complexes show a high intron/exon ratio, include loo** conformations between promoters and enhancers, and usually are developmentally regulated and/or tissue-specific. Multigene complexes display interactions among several promoters and often also include enhancers. The genes found in multigene complexes are shorter (i.e., with lower intron/exon ratio), more enriched in GC, and are located in highly transcribed, gene-dense euchromatin regions that are rich in short interspersed nuclear elements (SINEs). Recent genomic studies indicate that, on average, there are more than eight genes per multigene complex [61], suggesting that promoter-promoter aggregates are a major feature of eukaryotic gene regulation. Such complexes provide the topological basis for common transcriptional regulation of gene groups. For instance, the 58 HIST1H genes located on chromosome 6 are organized into three complexes that further interact to form a larger complex [61]. It is tempting to speculate that poised plasticity-related genes share common transcription factories enriched in the same transcriptional regulators. This could occur through promoter-promoter interactions, which could ultimately synchronize their rapid expression due to higher-order chromatin structures in which RNAPII acts as a primary hub.
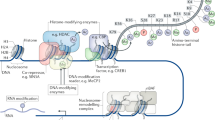
The activity of these transcription factories is dynamically regulated by the phosphorylation of specific serine (Ser) residues at the C-terminus domain (CTD) of RPB1, the largest subunit of the RNAPII complex [63]. Although the phosphorylation of the Ser5 is required for transcription initiation, RNAPII remains incapable of elongation because NELF binding pauses nascent RNA synthesis and stalls RNAPII downstream of the TSS. To unlock stalling and engage in productive elongation, it is necessary the phosphorylations of RPB1 at Ser2 and the pausing factors NELF and DRB sensitivity–inducing factor (DSIF). Both repressors, upon phosphorylation, turn into positive regulators [64]. Recent Chip-seq experiments have revealed over 8000 gene promoters on which the RNAPII is stalled [65]. This state is often referred to as ¨poised polymerase¨ and has been shown to be a common feature of the TSSs of immediate early genes (IEGs) in neurons, enabling their rapid transcriptional recruitment upon neuronal activity [65] (Fig. 2a). A mechanism reported to contribute to the attachment of IEGs to transcription factories is the de novo acetylation of SINEs located around their promoter (Fig. 2b). This process is controlled by TFIIIC, a general TF that represses IEG transcription in the basal state. As such, the depletion of the TFIIIC subunit Gtf3c5 enhances the localization of IEGs in transcription factories, and subsequently favors their transcription and promotes dendritogenesis [41]. How TFIIIC mediates this effect is yet unclear, although it has been hypothesized that the acetylation of SINEs could be mediated through either its TFIIIC90 subunit that has intrinsic lysine acetyltransferase (KAT) activity [66], or by recruiting coactivators such as p300 that have KAT activity [41]. Another regulatory mechanism of activity-driven transcription may rely on the appearance of DNA double-strand breaks (DSBs). Thus, it has been recently shown that DSBs and the phosphorylation of histone variant H2AX occur at specific genomic loci, including the TSSs of several IEGs, after neuronal stimulation (Fig. 2b). Two hours later, DSBs were repaired and transcription was back to basal levels [67]. Intriguingly, although the artificial induction of DSBs mostly caused gene downregulation, some IEGs exhibited the opposite response suggesting a physiological role for DSBs in productive elongation.

Activity-driven promoter/enhancer interactions leading to transcriptional elongation. a. In the basal state, RNAPII appears in transcriptional factories (an incompletely described proteinaceous body that is depicted in the scheme as a large blue globe) (1). The C-terminus of RPB1 has 52 tandem repeats of the heptapeptide YSPTSPS that contains two Ser residues that are dynamically phosphorylated. S5 phosphorylation (in orange) and the presence of the transcriptional repressors NELF and DSIF impede transcriptional elongation and stall RNAPII at gene promoters (2). b. Upon neuronal activity, distal enhancer sequences interact with the promoter thanks to the action of cohesin (3), which together with acetylated TFIIIC-bound SINEs mediates the relocation of plasticity genes. Enhancer acetylation requires the action of lysine acetyltransferases (4), such as CBP and p300, subsequently promoting their relocation. Transcriptional machinery (elongating RNAPII, the Mediator complex and TFs) binds to the enhancer element in order to transcribe eRNAs (5) that in turn bind to NELF and release it from the promoter. Finally, the phosphorylations of RNAPII (at Ser2), NELF and DSIF (red circles) would trigger productive elongation (6). In addition, it has been recently proposed that Topo IIB-mediated DSBs (upstream of the TSS) eliminate the loop that separates the promoter from the transcription factory (7)
Chromatin architecture in neuropsychiatric disease
As introduced in previous sections, the neurons in some developmental and degenerative disorders often display gross nuclear aberrations, while psychiatric disorders have been associated with more subtle changes (Table 1). We discuss below some additional examples that demonstrate the strong connection between aberrant chromatin architecture in neurons and neuropathology.
Neurodevelopmental disorders
Mutations in genes encoding proteins important for nuclear architecture (e.g. CTCF, cohesin and many epigenetic factors) frequently result in neurodevelopmental disorders [68]. This is the case of Opitz-Kaveggia syndrome and Fryns-Lujan syndrome which are both caused by mutations in MED12 [69] that encodes a subunit of the mediator complex. Moreover, mutations in the genes encoding either NIPBL or the cohesin subunits SMC1 and SMC3 cause Cornelia de Lange syndrome [70], whereas mutations in the CTCF gene have been associated with intellectual disability (ID), microcephaly and growth retardation [71]. Further supporting the link between aberrant chromatin structure and ID, various genes encoding proteins that interact with heterochromatin, such as ATRX and MeCP2, are also linked to ID. Thus, mutations in the gene that encodes ATRX cause Alpha-Thalassemia X-Linked ID syndrome [72], while the loss of MeCP2 results in Rett syndrome [73] that manifests itself with ID and autistic traits. Neurons lacking MeCP2 show an abnormal number and size of nucleoli and chromocenters [74], and an aberrant distribution of pericentric heterochromatinization [75]. Other syndromes are also characterized by nuclear defects even though their etiology is not directly linked to nuclear organizers. For instance, hippocampal neurons with CGG repeat expansions in the FMR1 gene, which give rise to fragile X-associated tremor/ataxia syndrome (FXTAS), accumulate more heterochromatin but in smaller foci [76].
Another type of genetic disorders associated with abnormal nuclear architecture are laminopathies in which the nuclear lamina is prominently disrupted. This group of disorders includes Hutchinson–Gilford progeria syndrome (HGPS) that is caused by mutations in the gene encoding lamin A [77]. Intriguingly, hippocampal nuclei of mouse models for this condition show abnormal lobulations and deep infoldings of the nuclear envelope, but gene expression and behavioral assays revealed no gross impairment [78], which indicates that neuronal nuclei can adapt to major perturbations in its structure. In contrast, as we will discuss in further detail for psychiatric conditions, other studies have shown that even local chromatin loo** perturbations might lead to neurological symptoms. For example, the single nucleotide polymorphism (SNP) rs12469063 associated with Restless Legs syndrome, a sensorimotor neurological disorder, has been shown to cause loo** perturbations and motor restlessness/hyperactivity in mouse models for this condition [79].
Neurodegenerative disorders
Large-scale chromatin reorganization is often observed in neurons undergoing degeneration. Thus, irregularities in nuclear shape, particularly mediated by B-type lamins, have been described to precede heterochromatin relaxation, DNA damage and neurodegeneration in both Drosophila models of tauopathy and human samples from Alzheimer’s patients [80]. Furthermore, dispersion of the nuclear lamina is known to precede neuronal death and is a common feature seen in mouse models of Alzheimer’s disease [81]. Other alterations may not cause prominent structural changes but still affect function. For example, mouse models for Huntington’s disease (HD) exhibit diminished super-enhancer function of striatum-specific genes governed by Gata2 and display reduced H3K27ac and paused RNAPII binding [82].
Psychiatric disorders
Aberrant chromatin loo**s have been recently implicated in psychiatric disorders. For example, Akbarian and colleagues first found that overexpression of the histone methyltransferase Setdb1 caused the heterochromatinization of the promoter of Grin2b (encoding for a subunit of the NMDA receptor) and the loss of a loop tethering the promoter to a Setdb1 target site positioned 30 kb downstream of the TSS [83]. Further investigation of the same locus revealed that the SNP rs117578877, located at the distal arm of another GRIN2B loop, is often found in schizophrenic patients and correlates with impaired working memory and schizotypic features. Notably, isogenic deletions of loop-bound sequences in mice impaired cognitive performance and decreased Grin2b expression [84]. The same team has also reported abnormal chromosomal interactions at a second locus linked to schizophrenia. The formation of a chromatin loop between the TSS of GAD1 (encoding an enzyme critical for GABA synthesis) and an enhancer sequence 50 Kb upstream was found reduced in the prefrontal cortex of schizophrenic patients [85]. A similar loop, sensitive to neuronal activation, was also detected in GABAergic neurons of mice. As a third example, it was recently demonstrated that a polymorphism affecting the interaction between the TSS of FKBP5, which encodes the co-chaperone FK506 binding protein 5, and enhancer sequences located in introns 2 and 7 is associated with an increased risk of develo** stress-related psychiatric disorders after childhood trauma [86]. Another recent study has shown that microsatellite repeats in intron 1 of the gene encoding neuregulin 1 (NRG1), a putative schizophrenia susceptibility gene regulating the excitatory-inhibitory balance, are associated with an increase in NRG1 transcripts in the prefrontal cortex, suggesting that this region could function as a transcriptional enhancer. Intriguingly, the presence of these repeats correlated with an earlier age of onset of the symptoms. However, long-range interactions between the intronic sequence and the promoter remain to be experimentally proven [87]. There are additional examples suggesting that abnormalities in chromatin loo** may be associated with conditions such a bipolar disorder [88] and impulsive-disinhibited personality [89], but molecular studies are still needed to prove the involvement of aberrant chromatin interactions in the etiology of these disorders.
Cause or consequence
Given the difficulty of examining the specific contribution of chromatin conformation changes through gain- and loss-of-function experiments, most of the evidence discussed above is correlative. A recent study by our team investigating transgenic mice that express high levels of GFP-tagged H2B in forebrain principal neurons has provided evidence for a causal role of aberrant chromatin organization in the emergence of neuropsychiatric traits [90]. Neuronal nuclei in these mice presented an aberrant subnuclear pattern resulting from chromocenter declustering, a loss of perinuclear heterochromatin, heterodense nucleoplasm, and abnormal distribution of heterochromatic and euchromatic epigenetic markers (Fig. 3). The mice also exhibited a number of phenotypes related to neuropsychiatric symptoms, such as hyperlocomotor activity, impaired social interactions, nociception, sensorimotor gating and memory, and the downregulation of several serotonin receptor genes that sit in the edge of “gene desert” zones [90]. Suggestively, this topographical feature is conserved in the human genome and might relate to the susceptibility of these loci to epigenetic deregulation. In addition to this work, the aforementioned studies conducted by the Akbarian’s lab on chromosomal loops at schizophrenia-linked genes further support a causal link between the loss of specific chromatin loops, transcriptional deregulation and neuronal alterations [83–85].

Chromatin perturbations cause behavioral impairments. The expression of the chimeric histone H2B-GFP causes dramatic changes in chromatin architecture, including the loss of peripheral heterochromatin, chromocenter declustering and changes in the texture of the nucleoplasm. This is likely due to stearic impediment of highly-packed tertiary chromatin fiber folding in heterochomatin by the protruding GFP tags. Remarkably, Htr1a alleles (red circles) relocated into the aberrant DNA foci, possibly explaining their downregulation and concomitant alterations in serotonin signaling and behavior
Excitingly, the use of engineered transcription factors has recently demonstrated that the local manipulation of epigenetic profiles at a given gene is sufficient to control drug- and stress-evoked transcriptional and behavioral responses, thereby providing seminal evidence for a causative role for those epigenetic marks [91]. Similarly, CRISPR/Cas9 technology now enables direct manipulation of genome topology, opening up the possibility to conduct loss- and gain-of-function experiments exploring the role of altered DNA conformations in pathology and transcription [84]. For example, CRISPR/Cas9 has recently been used to change the orientation of two interacting chromosomal regions, demonstrating that the functionality in vivo of some enhancers carrying CTCF-binding sites relies on their relative orientation and the precise architecture of chromatin domains [92].
Conclusions and prospects
As reviewed here, numerous studies have illustrated that nuclear architecture and genome topology are key for understanding neuronal function and dysfunction. Changes in subnuclear structures and chromatin loo**s have been found to occur in different neuronal plasticity paradigms. Similarly, the disruption of chromatin structures is a landmark for numerous neurological disorders. Although such a disruption likely contributes to the onset of a disorder, a clear distinction between cause and consequence is still missing, except for some monogenic disorders (often associated with ID) caused by mutations in architectural proteins or regulatory sequences. Although the specific contribution of architectural proteins and the changes in 3D chromatin organization to neuroplasticity and neuropathology largely remain to be determined, new light will soon be shed now that novel techniques such as super-resolution microscopy, NGS-based techniques for the analysis of DNA conformation and CRISPR/Cas9-based epi-editing have emerged. These innovative approaches will facilitate a high resolution determination of the 3D organization of the genome, in parallel to a systems-level interrogation of the consequences of gene expression, the identification of loci associated with aberrant function, and even the manipulation of DNA conformations to promote or correct transcriptional changes.
References
Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369(1633):20130288.
Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294(5544):1030–8.
Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80(3):624–32.
Wilczynski GM. Significance of higher-order chromatin architecture for neuronal function and dysfunction. Neuropharmacology. 2014;80:28–33.
Schoenfelder S, Clay I, Fraser P. The transcriptional interactome: gene expression in 3D. Curr Opin Genet Dev. 2010;20(2):127–33.
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–12.
Dekker J. Two ways to fold the genome during the cell cycle: insights obtained with chromosome conformation capture. Epigenetics Chromatin. 2014;7(1):25.
Aoto T, Saitoh N, Ichimura T, Niwa H, Nakao M. Nuclear and chromatin reorganization in the MHC-Oct3/4 locus at developmental phases of embryonic stem cell differentiation. Dev Biol. 2006;298(2):354–67.
Francastel C, Schübeler D, Martin DI, Groudine M. Nuclear compartmentalization and gene activity. Nat Rev Mol Cell Biol. 2000;1(2):137–43.
Berchtold D, Fesser S, Bachmann G, Kaiser A, Eilert J-C, Frohns F, Sadoni N, Muck J, Kremmer E, Eick D, Layer PG, Zink D. Nuclei of chicken neurons in tissues and three-dimensional cell cultures are organized into distinct radial zones. Chromosome Res. 2011;19(2):165–82.
Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol. 1999;145(6):1119–31.
Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, Joffe B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell. 2009;137(2):356–68.
Takizawa T, Meshorer E. Chromatin and nuclear architecture in the nervous system. Trends Neurosci. 2008;31(7):343–52.
Malhas A, Goulbourne C, Vaux DJ. The nucleoplasmic reticulum: form and function. Trends Cell Biol. 2011;21(6):362–73.
Wiegert JS, Bading H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium. 2011;49(5):296–305.
Wittmann M, Queisser G, Eder A, Wiegert JS, Bengtson CP, Hellwig A, Wittum G, Bading H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J Neurosci. 2009;29(47):14687–700.
Gruenbaum Y, Foisner R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu Rev Biochem. 2014. 84 (1):150306093657004–150306093657004.
Takamori Y, Tamura Y, Kataoka Y, Cui Y, Seo S, Kanazawa T, Kurokawa K, Yamada H. Differential expression of nuclear lamin, the major component of nuclear lamina, during neurogenesis in two germinal regions of adult rat brain. Eur J Neurosci. 2007;25(6):1653–62.
Lee JM, Tu Y, Tatar A, Wu D, Nobumori C, Jung H-J, Yoshinaga Y, Coffinier C, de Jong PJ, Fong LG, Young SG. Reciprocal knock-in mice to investigate the functional redundancy of lamin B1 and lamin B2. Mol Biol Cell. 2014;25(10):1666–75.
Jung H-J, Nobumori C, Goulbourne CN, Tu Y, Lee JM, Tatar A, Wu D, Yoshinaga Y, de Jong PJ, Coffinier C, Fong LG, Young SG. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc Natl Acad Sci U S A. 2013;110(21):E1923–32.
Zullo JM, Demarco IA, Piqué-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, Singh H. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149(7):1474–87.
Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, **e W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331–6.
Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38(4):603–13.
Tsai SY, Chuang JY, Tsai MS, Wang XF, ** ZX, Hung JJ, Chang WC, Bonci A, Su TP. Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc Natl Acad Sci U S A. 2015;112(47):E6562–70.
Alexander JM, Lomvardas S. Nuclear architecture as an epigenetic regulator of neural development and function. Neuroscience. 2014;264:39–50.
Jordan BA, Fernholz BD, Khatri L, Ziff EB. Activity-dependent AIDA-1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat Neurosci. 2007;10(4):427–35.
Allen KD, Gourov AV, Harte C, Gao P, Lee C, Sylvain D, Splett JM, Oxberry WC, van de Nes PS, Troy-Regier MJ, Wolk J, Alarcon JM, Hernández AI. Nucleolar integrity is required for the maintenance of long-term synaptic plasticity. PLoS One. 2014;9(8):e104364.
Hall MH, Magalska A, Malinowska M, Ruszczycki B, Czaban I, Patel S, Ambrożek-Latecka M, Zołocińska E, Broszkiewicz H, Parobczak K, Nair RR, Rylski M, Pawlak R, Bramham CR, Wilczyński GM. Localization and regulation of PML bodies in the adult mouse brain. Brain Struct Funct. 2016;221(5):2511–25.
Baltanás FC, Casafont I, Lafarga V, Weruaga E, Alonso JR, Berciano MT, Lafarga M. Purkinje cell degeneration in pcd mice reveals large scale chromatin reorganization and gene silencing linked to defective DNA repair. J Biol Chem. 2011;286(32):28287–302.
Woulfe J. Nuclear bodies in neurodegenerative disease. Biochim Biophys Acta. 2008;1783(11):2195–206.
Zhang Y, Wong C-H, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, Mulawadi FH, Sung W-K, Nicolis S, Ahituv N, Ruan Y, Wei C-L. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013;504(7479):306–10.
Ricci Maria A, Manzo C, García-Parajo MF, Lakadamyali M, Cosma Maria P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160(6):1145–58.
Risca VI, Greenleaf WJ. Unraveling the 3D genome: genomics tools for multiscale exploration. Trends Genet. 2015;31(7):357–72.
Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306–11.
Cattoni DI, Valeri A, Le Gall A, Nollmann M. A matter of scale: how emerging technologies are redefining our view of chromosome architecture. Trends Genet. 2015;31(8):454–64.
Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446(7138):882–7.
Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DC, Aitken S, **e SQ, Morris KJ, Itoh M, Kawaji H, Jaeger I, Hayashizaki Y, Carninci P, Forrest AR, Consortium F, Semple CA, Dostie J, Pombo A, Nicodemi M. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol. 2015;11(12):852.
Borden J, Manuelidis L. Movement of the X chromosome in epilepsy. Science. 1988;242(4886):1687–91.
Billia F, Baskys A, Carlen PL, De Boni U. Rearrangement of centromeric satellite DNA in hippocampal neurons exhibiting long-term potentiation. Brain Res Mol Brain Res. 1992;14(1–2):101–8.
Walczak A, Szczepankiewicz AA, Ruszczycki B, Magalska A, Zamlynska K, Dzwonek J, Wilczek E, Zybura-Broda K, Rylski M, Malinowska M, Dabrowski M, Szczepinska T, Pawlowski K, Pyskaty M, Wlodarczyk J, Szczerbal I, Switonski M, Cremer M, Wilczynski GM. Novel higher-order epigenetic regulation of the Bdnf gene upon seizures. J Neurosci. 2013;33(6):2507–11.
Crepaldi L, Policarpi C, Coatti A, Sherlock WT, Jongbloets BC, Down TA, Riccio A. Binding of TFIIIC to sine elements controls the relocation of activity-dependent neuronal genes to transcription factories. PLoS Genet. 2013;9(8), e1003699.
Williams RRE, Azuara V, Perry P, Sauer S, Dvorkina M, Jørgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, Fisher AG. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci. 2006;119(Pt 1):132–40.
Plank JL, Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell. 2014;55(1):5–14.
Vermunt MW, Reinink P, Korving J, de Bruijn E, Creyghton PM, Basak O, Geeven G, Toonen PW, Lansu N, Meunier C, van Heesch S, Clevers H, de Laat W, Cuppen E, Creyghton MP. Large-scale identification of coregulated enhancer networks in the adult human brain. Cell Rep. 2014;9(2):767–79.
Kieffer-Kwon K-R, Tang Z, Mathe E, Qian J, Sung M-H, Li G, Resch W, Baek S, Pruett N, Grøntved L, Vian L, Nelson S, Zare H, Hakim O, Reyon D, Yamane A, Nakahashi H, Kovalchuk AL, Zou J, Joung JK, Sartorelli V, Wei C-L, Ruan X, Hager GL, Ruan Y, Casellas R. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013;155(7):1507–20.
Fitzpatrick DJ, Ryan CJ, Shah N, Greene D, Molony C, Shields DC. Genome-wide epistatic expression quantitative trait loci discovery in four human tissues reveals the importance of local chromosomal interactions governing gene expression. BMC Genomics. 2015;16(1):109.
Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465(7295):182–7.
Schaukowitch K, Joo J-Y, Liu X, Watts Jonathan K, Martinez C, Kim T-K. Enhancer RNA Facilitates NELF Release from Immediate Early Genes. Mol Cell. 2014;56(1):29–42.
Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK-H, Harmin DA, Greenberg ME. Genome-wide identification and characterization of functional neuronal activity–dependent enhancers. Nat Neurosci. 2014;17(10):1330–9.
Joo JY, Schaukowitch K, Farbiak L, Kilaru G, Kim TK. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci. 2016;19(1):75–83.
Burgess-Beusse B, Farrell C, Gaszner M, Litt M, Mutskov V, Recillas-Targa F, Simpson M, West A, Felsenfeld G. The insulation of genes from external enhancers and silencing chromatin. Proc Natl Acad Sci U S A. 2002;99 Suppl 4:16433–7.
Dowen Jill M, Fan Zi P, Hnisz D, Ren G, Abraham Brian J, Zhang Lyndon N, Weintraub Abraham S, Schuijers J, Lee Tong I, Zhao K, Young Richard A. Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes. Cell. 2014;159(2):374–87.
Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153(6):1281–95.
Cuadrado A, Remeseiro S, Graña O, Pisano DG, Losada A. The contribution of cohesin-SA1 to gene expression and chromatin architecture in two murine tissues. Nucleic Acids Res. 2015;43(6):3056–67.
Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, ** JLH, Mulawadi F, Wong E, Sheng J, Zhang Y, Poh T, Chan CS, Kunarso G, Shahab A, Bourque G, Cacheux-Rataboul V, Sung W-K, Ruan Y, Wei C-L. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43(7):630–8.
Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–5.
Watson LA, Wang X, Elbert A, Kernohan KD, Galjart N, Bérubé NG. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J Neurosci. 2014;34(8):2860–70.
Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2(2):345–57.
Melnik S, Deng B, Papantonis A, Baboo S, Carr IM, Cook PR. The proteomes of transcription factories containing RNA polymerases I, II or III. Nat Methods. 2011;8(11):963–8.
Edelman LB, Fraser P. Transcription factories: genetic programming in three dimensions. Curr Opin Genet Dev. 2012;22(2):110–4.
Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei C-L, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung W-K, Snyder M, Ruan Y. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148(1–2):84–98.
Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M, Taipale J. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154(4):801–13.
Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, Darzacq X. Real-time dynamics of RNA polymerase II clustering in live human cells. Science (New York, NY). 2013;341(6146):664–7.
Kwak H, Lis JT. Control of transcriptional elongation. Annu Rev Genet. 2013;47:483–508.
Saha RN, Wissink EM, Bailey ER, Zhao M, Fargo DC, Hwang JY, Daigle KR, Fenn JD, Adelman K, Dudek SM. Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat Neurosci. 2011;14(7):848–56.
Hsieh YJ, Kundu TK, Wang Z, Kovelman R, Roeder RG. The TFIIIC90 subunit of TFIIIC interacts with multiple components of the RNA polymerase III machinery and contains a histone-specific acetyltransferase activity. Mol Cell Biol. 1999;19(11):7697–704.
Madabhushi R, Gao F, Pfenning Andreas R, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao P-C, Stott Ryan T, Gjoneska E, Nott A, Cho S, Kellis M, Tsai L-H. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161(7):1592–605.
Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25(10):1473–81.
Philibert RA, Madan A. Role of MED12 in transcription and human behavior. Pharmacogenomics. 2007;8(8):909–16.
Remeseiro S, Cuadrado A, Kawauchi S, Calof AL, Lander AD, Losada A. Reduction of Nipbl impairs cohesin loading locally and affects transcription but not cohesion-dependent functions in a mouse model of Cornelia de Lange Syndrome. Biochim Biophys Acta. 2013;1832(12):2097–102.
Gregor A, Oti M, Kouwenhoven EN, Hoyer J, Sticht H, Ekici AB, Kjaergaard S, Rauch A, Stunnenberg HG, Uebe S, Vasileiou G, Reis A, Zhou H, Zweier C. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet. 2013;93(1):124–31.
Ratnakumar K, Bernstein E. ATRX: the case of a peculiar chromatin remodeler. Epigenetics. 2013;8(1):3–9.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8.
Singleton MK, Gonzales ML, Leung KN, Yasui DH, Schroeder DI, Dunaway K, LaSalle JM. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol Dis. 2011;43(1):190–200.
Agarwal N, Becker A, Jost KL, Haase S, Thakur BK, Brero A, Hardt T, Kudo S, Leonhardt H, Cardoso MC. MeCP2 Rett mutations affect large scale chromatin organization. Hum Mol Genet. 2011;20(21):4187–95.
Chen Y, Tassone F, Berman RF, Hagerman PJ, Hagerman RJ, Willemsen R, Pessah IN. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010;19(1):196–208.
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–8.
Baek J-H, Schmidt E, Viceconte N, Strandgren C, Pernold K, Richard TJC, Van Leeuwen FW, Dantuma NP, Damberg P, Hultenby K, Ulfhake B, Mugnaini E, Rozell B, Eriksson M. Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum Mol Genet. 2015;24(5):1305–21.
Spieler D, Kaffe M, Knauf F, Bessa J, Tena JJ, Giesert F, Schormair B, Tilch E, Lee H, Horsch M, Czamara D, Karbalai N, von Toerne C, Waldenberger M, Gieger C, Lichtner P, Claussnitzer M, Naumann R, Müller-Myhsok B, Torres M, Garrett L, Rozman J, Klingenspor M, Gailus-Durner V, Fuchs H, Hrabě de Angelis M, Beckers J, Hölter SM, Meitinger T, Hauck SM, Laumen H, Wurst W, Casares F, Gómez-Skarmeta JL, Winkelmann J. Restless legs syndrome-associated intronic common variant in Meis1 alters enhancer function in the develo** telencephalon. Genome Res. 2014;24(4):592–603.
Frost B, Bardai FH, Feany MB. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr Biol. 2016;26(1):129–36.
Chang K-H, Multani PS, Sun K-H, Vincent F, de Pablo Y, Ghosh S, Gupta R, Lee H-P, Lee H-G, Smith MA, Shah K. Nuclear envelope dispersion triggered by deregulated Cdk5 precedes neuronal death. Mol Biol Cell. 2011;22(9):1452–62.
Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, Neri C, Davidson I, Merienne K. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum Mol Genet. 2015;24(12):3481–96.
Jiang Y, Jakovcevski M, Bharadwaj R, Connor C, Schroeder FA, Lin CL, Straubhaar J, Martin G, Akbarian S. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30(21):7152–67.
Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY, Mitchell AC, Mao W, Whittle C, Dincer A, Jakovcevski M, Pothula V, Rasmussen TP, Giakoumaki SG, Bitsios P, Sherif A, Gardner PD, Ernst P, Ghose S, Sklar P, Haroutunian V, Tamminga C, Myers RH, Futai K, Wood MA, Akbarian S. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron. 2014;84(5):997–1008.
Bharadwaj R, Jiang Y, Mao W, Jakovcevski M, Dincer A, Krueger W, Garbett K, Whittle C, Tushir JS, Liu J, Sequeira A, Vawter MP, Gardner PD, Casaccia P, Rasmussen T, Bunney Jr WE, Mirnics K, Futai K, Akbarian S. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33(29):11839–51.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TWW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16(1):33–41.
Weickert CS, Tiwari Y, Schofield PR, Mowry BJ, Fullerton JM. Schizophrenia-associated HapICE haplotype is associated with increased NRG1 type III expression and high nucleotide diversity. Transl Psychiatry. 2012;2:e104.
Pedrosa E, Stefanescu R, Margolis B, Petruolo O, Lo Y, Nolan K, Novak T, Stopkova P, Lachman HM. Analysis of protocadherin alpha gene enhancer polymorphism in bipolar disorder and schizophrenia. Schizophr Res. 2008;102(1–3):210–9.
Laplana M, Royo JL, García LF, Aluja A, Gomez-Skarmeta JL, Fibla J. SIRPB1 copy-number polymorphism as candidate quantitative trait locus for impulsive-disinhibited personality. Genes Brain Behav. 2014;13(7):653–62.
Ito S, Magalska A, Alcaraz-Iborra M, Lopez-Atalaya JP, Rovira V, Contreras-Moreira B, Lipinski M, Olivares R, Martinez-Hernandez J, Ruszczycki B, Lujan R, Geijo-Barrientos E, Wilczynski GM, Barco A. Loss of neuronal 3D chromatin organization causes transcriptional and behavioural deficits related to serotonergic dysfunction. Nat Commun. 2014;5:4450.
Heller EA, Cates HM, Pena CJ, Sun H, Shao N, Feng J, Golden SA, Herman JP, Walsh JJ, Mazei-Robison M, Ferguson D, Knight S, Gerber MA, Nievera C, Han MH, Russo SJ, Tamminga CS, Neve RL, Shen L, Zhang HS, Zhang F, Nestler EJ. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–7.
Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, Li W, Zhang MQ, Ren B, Krainer AR, Maniatis T, Wu Q. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162(4):900–10.
Zannas AS, Binder EB. Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav. 2014;13(1):25–37.
Holz NE, Buchmann AF, Boecker R, Blomeyer D, Baumeister S, Wolf I, Rietschel M, Witt SH, Plichta MM, Meyer-Lindenberg A, Banaschewski T, Brandeis D, Laucht M. Role of FKBP5 in emotion processing: results on amygdala activity, connectivity and volume. Brain Struct Funct. 2015;220(3):1355–68.
Acknowledgments
We thank Grzegorz Wilcynski and members of Barco’s lab for critical reading of the manuscript.
Funding
AM is recipient of a Formación de Personal Investigador fellowship from the Spanish Ministry of Economy and Competitivity (MINECO). Research in Barco’s lab is supported by grants SAF2014-56197-R, PCIN-2015-192-C02-01 and SEV-2013-0317 from MINECO, grant PROMETEO/2016/006 from the Generalitat Valenciana, a NARSAD Independent Investigator Grant from the Brain & Behavior Research Foundation and a grant from the Alicia Koplowitz Foundation. The Instituto de Neurociencias is a “Centre of Excellence Severo Ochoa”.
Availability of data and materials
Not applicable.
Authors’ contributions
AM elaborated the figures, AM and AB wrote and revised the text. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Medrano-Fernández, A., Barco, A. Nuclear organization and 3D chromatin architecture in cognition and neuropsychiatric disorders. Mol Brain 9, 83 (2016). https://doi.org/10.1186/s13041-016-0263-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-016-0263-x