
Abstract
Background
Cerium (Ce) is a rare earth element, rapidly oxidizing to form CeO2, and currently used in numerous commercial applications, especially as nanoparticles (NP). The potential health effects of Ce remain uncertain, but literature indicates the development of rare earth pneumoconiosis accompanied with granuloma formation, interstitial fibrosis and inflammation. The exact underlying mechanisms are not yet completely understood, and we propose that autophagy could be an interesting target to study, particularly in macrophages. Therefore, the objective of our study was to investigate the role of macrophagic autophagy after pulmonary exposure to CeO2 NP in mice. Mice lacking the early autophagy gene Atg5 in their myeloid lineage and their wildtype counterparts were exposed to CeO2 NP by single oropharyngeal administration and sacrificed up to 1 month after. At that time, lung remodeling was thoroughly characterized (inflammatory cells infiltration, expression of fibrotic markers such as αSMA, TGFβ1, total and type I and III collagen deposition), as well as macrophage infiltration (quantification and M1/M2 phenotype).
Results
Such pulmonary exposure to CeO2 NP induces a progressive and dose-dependent lung fibrosis in the bronchiolar and alveolar walls, together with the activation of autophagy. Blockage of macrophagic autophagy protects from alveolar but not bronchiolar fibrosis, via the modulation of macrophage polarization towards M2 phenotype.
Conclusion
In conclusion, our findings bring novel insight on the role of macrophagic autophagy in lung fibrogenesis, and add to the current awareness of pulmonary macrophages as important players in the disease.
Similar content being viewed by others


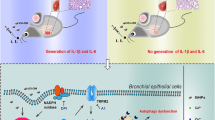
Background
Until recent years, rare earth elements (REE) received limited attention from environmental and public health researchers. In the last two decades, they have however undergone a fantastic boost in their technological and industrial utilization, which is accompanied by concerns regarding emissions and potential human exposures. One of the main REE currently used in numerous commercial applications is Cerium (Ce), as Ce is very reactive and is a strong oxidizing agent, rapidly oxidizing when in contact with oxygen to form CeO2. Ce is for example used in flat screen display, alloys, petroleum refining (cracking catalyst), ceramics, glass additives, phosphors, or polishing compounds for glass mirrors, plate glass, television tubes, ophthalmic lenses, precision optics, electronic wafers [1]. CeO2 is also used as additive in cigarettes, and CeO2 nanoparticles (NP) are predominantly used as diesel fuel additive to increase fuel combustion efficiency [2]. Because of these already numerous current and foreseen applications, CeO2 NP might enter the environment through disposal of consumer and industrial products, and this is accompanied by a growing concern for human health.
The potential health effects of Ce remain uncertain, but RE pneumoconiosis accompanied with granuloma and interstitial fibrosis has been reported in workers exposed to asbestos and RE dusts, together with the presence of Ce-containing particles in macrophages from bronchoalveolar lavage (BAL) and in interstitial macrophages [3, 4]. Experimental studies conducted in mice or rats have shown that animal exposure to CeO2 NP leads to the development of pulmonary fibrosis, accompanied by a persistent pulmonary inflammation and the presence of oxidative stress [1, 5,6,7,8,9]. The exact underlying mechanisms are not yet completely understood, and we propose that autophagy could be an interesting target to study.
Autophagy is a physiological process mainly responsible for the recycling of damaged cellular organelles and/or macromolecules into simpler forms for cell survival. The resulting degradation products are recycled to maintain nutrient and energy homeostasis [10, 11]. Apart from its importance in physiological conditions, numerous studies have suggested that autophagy is regulated under different pathophysiological conditions, in particular in the lung fibrotic process [12, 13]. We have recently demonstrated that macrophagic autophagy can be modulated after exposure to NP [14], which could be of relevance in the context of pulmonary response to Ce exposure given that macrophages are important players in fibrotic disease [15,16,17].
Therefore, the objective of the present study was to investigate the role of macrophagic autophagy after pulmonary exposure to CeO2 NP in mice. To achieve our aim, mice lacking the early autophagy gene Atg5 in their myeloid lineage and their wildtype counterparts were exposed to CeO2 NP by single oropharyngeal administration and sacrificed up to 1 month after. At that time, lung remodeling was thoroughly characterized (inflammatory cells infiltration, expression of fibrotic markers such as αSMA, TGFβ1, total and type I and III collagen deposition), as well as macrophage infiltration (quantification and M1/M2 phenotype).
Results
Characterization of pulmonary remodeling and autophagy modulation in response to CeO2 NP
Exposure of C57Bl/6 mice to a single administration of 50 μg CeO2 NP induced a profound lung fibrotic remodeling characterized by a progressive thickening of bronchiolar and alveolar walls. These modifications appeared gradually, starting from 24 h post CeO2 NP administration for bronchiolar modifications, and after 1 week for the alveolar ones (Fig. 1a-c). CeO2 NP were internalized in macrophages present in the alveolar area (Fig. 1d). This was accompanied by a persistent increased total cell count in the BAL (Fig. 2a). Cell differential analysis identified macrophages as the predominant cell population, with a transient significant increase of neutrophil percentage in BAL from CeO2-exposed animal 24 h and 1 week after their initial exposure, but which was no longer present after 1 month (Additional Figure 1). As shown in Fig. 2, CeO2 NP were internalized in BAL macrophages at all time points.

Exposure to CeO2 NP induces a progressive lung remodeling. Representative lung tissue sections of C57Bl/6 mice exposed to Saline or CeO2 NP and stained with Hematoxylin Eosin (HE) after 1 day (Panel a), 7 days (Panel b) or 28 days (Panel c). Original magnification × 200. Panel d: higher magnification of alveolar macrophages with internalized NP

Exposure to CeO2 NP induces an increase in BAL cellularity. Quantification of total cell count in BAL fluid (Panel a). Each individual circle represents the value obtained from one animal (empty circle: saline exposure – plain circle: CeO2 NP-exposure). *p < 0.05. Typical images of BAL macrophages from Saline-exposed (Panel b) or CeO2 NP-exposed (Panel c) mice sacrifice after 24 h, 1 week or 28 days
To investigate whether autophagy was induced in our experimental model of lung fibrosis, we utilized GFP-LC3 transgenic mice and exposed them to 50 μg CeO2 NP. As shown in Fig. 3, we were able to observe the induction of autophagy in CeO2-exposed GFP-LC3 mice, already 24 h after the initial administration, and up to 28 days. This was observed mainly in epithelial cells in bronchiolar regions, and in alveolar regions (Fig. 3). These results were confirmed by the observation of increased levels of Atg5 protein, an early marker of autophagy pathway, in similar regions of CeO2-exposed mice (Fig. 3d).

Exposure to CeO2 NP induces autophagy. Representative lung tissue sections of GFP-LC3 mice exposed to Saline or CeO2 NP after 1 day (Panel a), 7 days (Panel b) or 28 days (Panel c). Original magnification × 200. Panel d: representative lung tissue sections of C57Bl/6 mice exposed to Saline or 50 μg CeO2 NP and stained, after 28 days, for Atg5. Original magnification × 200
As macrophages are supposedly important players in lung fibrosis [15,16,17,18], and as we recently demonstrated the importance of autophagy in macrophagic response to NP [14], we next focus our work on macrophages. The potential of CeO2 NP to induce autophagy in macrophages was confirmed by the observed accumulation of GFP-LC3 puncta in peritoneal macrophages obtained from GFP-LC3 mice and exposed to CeO2 NP in vitro (Additional Figure 2A), as well as by the induction of Atg5 protein expression in peritoneal macrophages of C57Bl/6 mice exposed to CeO2 NP in vitro (Additional Figure 2B). Finally, as autophagy is a highly dynamic process, we addressed the issue of its activation in macrophages exposed to CeO2 NP. As shown in Additional Figure 2C, we could observe the colocalization of LC3 staining with that of the lysosomal protein LAMP-1 in CeO2 NP-exposed macrophages. Overall, these results indicate that pulmonary exposure to CeO2 NP is followed by the activation of autophagy, including in macrophages, which are able to phagocyte CeO2 NP.
Effect of blockage of macrophagic autophagy in response to CeO2 NP
In order to decipher the specific role of macrophagic autophagy in the development of lung fibrosis, mice lacking Atg5 gene in their myeloid lineage (Atg5flox/flox LysMCre or Atg5+/−) were exposed to CeO2 NP. Moreover, to further characterize our model, we performed a dose-response experiment and analyzed the lung remodeling achieved in mice 28 days after exposure to one single administration of either 5 or 50 μg CeO2 NP.
As shown in Figs. 4 & 5, Atg5+/− mice do not present any particular lung phenotype in absence of exogenous exposure (Saline condition), and their macrophages do not express Atg5 protein in response to CeO2 NP (Additional Figure 3). As for their WT counterparts, no major histological modification could be observed 28 days after the administration of 5 μg CeO2 NP (Figs. 4 & 5). However, when considering the higher dose of 50 μg CeO2 NP, histological analysis of lung tissue sections indicates that Atg5+/− mice were protected against CeO2 NP-induced alveolar fibrosis; no alveolar wall thickening could be observed in these mice (Fig. 4 *: p < 0.05). This protection against alveolar remodeling was further confirmed by the quantification of total, type I and III collagen deposition, as well as expression of αSMA and TGF-ß: significant differences between Atg5+/− and WT mice could be detected for these parameters. In stark contrast, Atg5+/− mice present the same peribronchiolar fibrosis as compared to their WT counterparts (Fig. 5, *: p < 0.05); a significant thickening of bronchiolar walls, together with a significant accumulation of total collagen, collagen I, collagen III, αSMA and TGF-ß in bronchiolar area could be observed in Atg5+/− mice exposed to 50 μg CeO2 NP, and was not different between Atg5+/− and WT mice.

Atg5+/− mice are protected from alveolar remodeling. Representative lung tissue sections of WT and Atg5+/− mice exposed to Saline or CeO2 NP (5 or 50 μg, observation at 28 days), after HES (Panel a and b for quantification), Sirius red (Panel c and d for quantification), Collagen type I (Panel e and f for quantification), Collagen type III (Panel g and h for quantification), αSMA (Panel i and j for quantification) and TGF-ß (Panel k and l for quantification). Original magnification × 200. Each individual circle represents the mean value obtained from one animal. Empty circle: saline exposure. Grey circle: 5 μg CeO2 NP exposure. Plain circle: 50 μg CeO2 NP exposure. *p < 0.05

Atg5+/− mice are not protected from bronchiolar remodeling. Representative lung tissue sections of WT and Atg5+/− mice exposed to Saline or CeO2 NP (5 or 50 μg, observation at 28 days), after HES (Panel a and b for quantification), Sirius red (Panel c and d for quantification), Collagen type I (Panel e and f for quantification), Collagen type III (Panel g and h for quantification), αSMA (Panel i and j for quantification) and TGF-ß (Panel k and l for quantification). Original magnification × 200. Each individual circle represents the mean value obtained from one animal. Empty circle: saline exposure. Grey circle: 5 μg CeO2 NP exposure. Plain circle: 50 μg CeO2 NP exposure. *p < 0.05
This protection wasn’t the result of a differential CeO2 NP accumulation between WT and Atg5+/− mice, as X-Ray microfluorescence (micro-XRF) showed that the Ce signal could be observed with similar intensity whatever the mice genotype, mainly in the alveolar region, and much less in bronchiolar walls (Additional Figure 4). Moreover, micro X-ray absorption near edge structure (XANES) spectra of Ce spots detected in CeO2-exposed mice revealed three different types of modifications that could be observed: 1/ similar to that of the CeO2 reference (red line in Additional Figure 4E – Spectrum Type I); 2/ a spectrum presenting a shift of the first peak toward 5727 eV (instead of 5731 eV as in the reference – yellow line – Spectrum Type II), suggesting the reduction of CeIV to CeIII; and 3/ a mixed contribution of Type I and Type II spectra (green line). A similar distribution of the different type of spectra could be observed in all CeO2-exposed lungs, whatever the genotype (Table 1).
Characterization of macrophage polarization in response to CeO2 NP
Blockage of macrophagic autophagy didn’t modify the increase in total number of macrophages induced by 50 μg CeO2 NP exposure (Fig. 6), while no modification of total macrophage number could be observed in response to 5 μg CeO2 NP, whatever the mouse genotype. Indeed, the quantification of CD107b expression in lung tissue sections of CeO2-exposed mice, as an index of total number of macrophages in lung tissue, was similar in WT and Atg5+/− mice, and confirmed the increased total number of macrophages observed in BAL (Fig. 6a and b). However, when assessing the M1 or M2 phenotype of alveolar macrophages as potential contributors to alveolar fibrotic response [15,16,17], we observed that WT mice exposed to 50 μg CeO2 NP (but not 5 μg) present an M1-like phenotype, with significantly increased expression of CD68, CD80 and iNOS proteins as compared to unexposed mice (Fig. 6c-e and i-k, *: p < 0.005), whereas Atg5+/− mice developed an M2-like phenotype, characterized by increased expression of CD206, CD163 and Arginase1 (Fig. 6f-h and l-n,*: p < 0.005). These results were confirmed in vitro; exposure of WT peritoneal macrophages to CeO2 NP led to the increased expression of M1 markers, while exposure of macrophages obtained from Atg5+/− mice lead to an increased expression of M2 markers (Additional Figure 5).

Characterization of CeO2-induced macrophage recruitment and polarization in mice. Representative lung tissue sections of C57Bl/6 mice exposed to Saline or CeO2 NP (5 or 50 μg, observation at 28 days), after immunostaining for CD107b, as a marker of total number of macrophages (Panel a). Original magnification × 200. Quantification of CD107b positive cells (Panel b). Representative lung tissue sections of mice exposed to Saline or CeO2 NP (5 or 50 μg, observation at 28 days), after immunostaining for CD80 (Panel c and d for quantification), CD68 (Panel e and f for quantification), iNOS (Panel g and h for quantification), CD206 (Panel i and j for quantification), CD163 (Panel k and l for quantification) or Arginase 1 (Panel m and n for quantification). Original magnification × 200. Legends as in Fig. 4
This modification of macrophage polarization in Atg5+/− mice could be responsible for the protective effect against the development of alveolar fibrosis in Atg5+/− mice. Indeed, exposure of WT fibroblasts to the supernatant of CeO2-exposed macrophages induced fibroblast-to-myofibroblast differentiation, detected by an increased expression of αSMA, Collagen I and III, only when WT macrophages were used, but not macrophages bearing a Atg5+/− genotype (Fig. 7).

Fibroblast differentiation in vitro. Representative images of WT fibroblasts exposed to supernatant of Vehicle (Control) or CeO2 NP-exposed macrophages obtained from WT or Atg5+/− mice, and immuno-stained for SMA, Collagen type I or type III
Discussion
Taken together, our results show the progressive and dose-dependent development of lung fibrosis in mice exposed to CeO2 NP. They also demonstrate that the blockage of macrophagic autophagy protects from alveolar but not bronchiolar fibrosis, probably via the modulation of macrophage polarization in favor of a M2 phenotype.
The progressive and dose-dependent induction of pulmonary fibrosis that we describe here in response to CeO2 NP single administration is in accordance with data from literature using other CeO2 NP [1, 5,6,7,8,9]. The same is true for the sustained pulmonary inflammation that we detected and which has been shown to be slow to resolve post exposure [19]. We didn’t however observe the formation of granuloma as described for example by Park and colleagues in CeO2-exposed animals [8], but this is probably linked to the lower doses of CeO2 NP that we used in our study; 5-50 μg per mouse versus 3 mg in Park study. These doses were chosen based on the effects seen in other studies, covering a range from minimal to clearly observed effects. Our results also confirm and extend the data obtained by Ma and colleagues, showing that rats exposure to CeO2 NP led to the induction of M1 phenotype in BAL macrophages of these animals [20]. It must be noted however that 28 days after the initial administration, mRNA expression of Arginase 1 was increased in Park study, thus suggesting a shift toward M2 phenotype that was not observed in our WT animals, although care must be taken as only one single M2 marker was targeted in Park’s work.
The development of bronchiolar fibrosis in response to CeO2 NP was not prevented by the blockage of macrophagic autophagy. Interestingly, this could be paralleled by the very low Ce elemental signal observed by μXRF in bronchiolar regions, as well as the almost complete lack of macrophages present in these areas, suggesting that CeO2-induced bronchiolar fibrosis is independent of macrophages. Although we did not investigate further the biological mechanism(s) underlying this bronchiolar fibrosis, the occurrence of epithelial-mesenchymal transition (EMT) could be an explanation. Indeed, Ma and colleagues have recently demonstrated that exposure to CeO2 NP induces EMT in alveolar type II cells that ultimately plays a role in lung fibrosis [5]. Similarly to what we have demonstrated with in vitro exposure of fibroblasts to the supernatant of Ce-exposed macrophages, it could be interesting to explore the effect of the secretome from Ce-exposed epithelial cells on fibroblast to myofibroblast differentiation in our system.
The protection against alveolar fibrogenesis observed in Ce-exposed Atg5+/− mice could have been the result of decreased amounts of Ce present in alveolar regions of Atg5+/− mice. Indeed, autophagy is known to interplay with macrophage phagocytosis [21,22,23], and M2 polarization has been shown to enhance Si-NP uptake by macrophages [24]. Although we did not strictly quantify the Ce content in WT and Atg5+/− mice exposed to CeO2 NP, the similar μXRF signals obtained 28 days after the initial NP administration in both animal genotypes in terms of both intensity and tissue distribution, together with the similar total number of macrophages present in the lungs of WT and Atg5+/− animals strongly suggest that the protection against alveolar fibrosis observed in Atg5+/− animals is probably not the result of a decreased amount of CeO2 NP in these individuals. It must be added, however, that the quantification of the initial deposition of NP in the first 24 h after their instillation should deserve further studies in order to fully understand the following fibrotic response. CeO2 is highly reactive, and it has been suggested that a change in the Ce3+/Ce4+ ratio may play a significant role in toxicity determination [25], and could thus contribute to lung fibrosis development. In our experiment, while Ce in CeO2 NP was Ce4+, Ce in lung tissue was only 33–36% pure Ce4+, indicating a change in speciation after NP administration. However, both WT and Atg5+/− mice showed a similar modification of their Ce speciation, with 45–48% as Ce3+, and around 18% presenting a mix Ce3+/Ce4+ form. Therefore, the specific protection against alveolar fibrosis observed in Atg5+/− mice could not be attributed to modifications of Ce speciation.
The modification of macrophage polarization related to their autophagy status could also represent an interesting candidate to explore the underlying mechanism that occurs in Atg5+/− mice [26]. Indeed, macrophages are divided into two distinct sub-populations defined as classically activated pro-inflammatory M1 subtype and alternatively activated M2 subtype responsible for anti-inflammatory, tissue repair and remodeling [27]. Both M1 and M2 macrophages have been noted to be involved in the pathogenesis of pulmonary fibrosis. This highlights the plasticity of macrophage polarization depending on the micro-environment stimuli and signals [28], as well as the fact that assays dedicated to polarization generally capture what happens at a given time point in time and space, as we did in our study, while a wiser approach could have been to consider the timing of the modifications in polarization [29]. In our experimental model of lung fibrosis, we demonstrated that macrophages from autophagy-deficient mice tended to polarize into M2 phenotype whereas their wild-type counterparts exhibiting proficient autophagy skewed towards the M1 subtype. Interestingly, the induction of autophagy by advanced glycation end products or rapamycin triggered macrophage polarization toward M1 phenotype, as well as a sustained inflammation in mice and patients, resulting in delayed wound healing. Moreover, the inhibition of autophagy reduced M1 population, which is in accordance with our results [30]. However, it must be noted that in mice on a high fat diet, there was an increased M1 and decreased M2 polarization in macrophages with defective autophagy, leading to hepatic inflammation and the progression to liver injury [31]. This underlines the importance of the environmental context in the overall autophagy effect, which could be a clue to the effects observed in our study, as to understand the underlying mechanism of macrophage specific polarization in absence of autophagy.
Finally, our results suggest that macrophagic autophagy can facilitate fibrosis. As stated before, the induction of lung fibrosis by CeO2 NP is in accordance with experimental data from literature, exploring the consequences of pulmonary administration of CeO2 NP but also chemically different NP, such as Silica (Si) NP [1, 5,6,7,8,9, 32]. When it comes to the role of autophagy in this particle-induced lung fibrotic process, the literature is far less clear [33], as it contains both references that autophagy is protective [11, 13, 34, 35], and also that it can be deleterious [36,37,38,39]. Using the same mice model, Jessop and colleagues demonstrated a greater silica-induced lung fibrosis, together with enhanced inflammation in Atg5+/− mice as compared to their WT littermates [40]. This result, which is opposite to ours, could be explained by the chemical nature of the particles used (silica versus cerium), together with their size (micro versus nano) as these factors are known to interfere with the resulting effect on autophagy process [41]. Moreover, an important difference between the two studies could be the toxicity which was observed in response to silica but not cerium oxide particles exposure, toxicity that could ultimately lead to the modulation of alternative mechanisms of action linked to fibrosis pathophysiology, such as NLRP3 inflammasome [40, 42], IL-1ß signaling [43, 44], EMT [34], macrophage polarization [38, 45], senescence [37], lipid metabolism [46], or apoptosis [47], or endoplasmic reticulum stress [48]. Recent results from the literature show an increased number of LC3-II puncta, an index of active autophagy, in fibroblast foci of patients with idiopathic pulmonary fibrosis (IPF), the most common form of pulmonary fibrosis [13, 39], which is in accordance with our results, although one can’t know if it results from an effort to protect rather than contribute to the disease. In vitro, these authors also showed that autophagy is induced by TGFß, and that it is necessary for TGFß-induced fibrosis in both non-IPF and IPF fibroblasts [7]. Interestingly, such a cell-specific role for autophagy has been recently described in chronic obstructive pulmonary disease (COPD) where autophagy is believed to be protective except in macrophages [10,11,12, 34]. In lung fibrosis, the majority of the studies have focused on lung epithelial cells and/or fibroblasts [34], as the two major cell types involved in fibrogenesis [18]. However, the recruitment of inflammatory cells, leading to the activation of effector cells, is often considered as a first trigger of fibrosis [49], and as such, the activation of macrophages occupies a pivotal role in the translation of injury to aberrant repair in lung fibrosis [16, 17]. Interestingly, the LC3 puncta observed by Ghavami and colleagues in fibroblast foci of IPF patients are coherent with the presence of macrophages [39].
Overall, our work provides in vivo as well as in vitro evidences of the role of macrophagic autophagy in CeO2 NP-induced lung alveolar fibrosis. We are fully aware that peritoneal macrophages do not fully represent the lung resident macrophages or alveolar macrophages. However, we chose to use them because a large body of literature combining both in vitro approaches using peritoneal macrophages, and in vivo experiments in mice, demonstrates the relevance of the findings obtained in peritoneal macrophages, and their potential translation to what may occur in vivo [50,51,52,53]. Moreover, we wanted to be at most in accordance with the 3R’s guidelines, as the number of alveolar macrophages that can be obtained from a broncho-alveolar lavage performed in one single mouse would have required a very high number of animals to obtain a sufficient amount of cells for in vitro experiments. We therefore strongly believe that our in vitro results are relevant to what occurs in vivo, although this specific issue should be addressed by dedicated studies that were beyond the scope of the present study.
Conclusions
In conclusion, although we are perfectly conscious that one single administration of CeO2 NP doesn’t recapitulate the entire human development of fibrotic disease our findings bring novel insight on the role of macrophagic autophagy in lung fibrogenesis, and add to the current awareness of pulmonary macrophages as important players in the disease.
Methods
Experimental model of lung fibrosis
Eight to twelve weeks old C57Bl/6 mice purchased from Janvier (Le Genest-St-Isle, France) were acclimated for 1 week. All mice (maximum 5 per cage) were supplied with food (SAFE, Auguy, France) and tap sterilized water ad libitum in standard wire-topped cages in a controlled environment, with a 12 h light/dark cycle. They then received saline or CeO2 NP (5 or 50 μg, NanoAmor, Houston, TX) via non-surgical oropharyngeal instillation (MicroSprayer® Aerosolizer, PennCentury), using the intubation platform and small animal laryngoscope (Penn-Century, Inc) for holding the mice. Before each oropharyngeal instillation, mice have been beforehand anesthetized using intro-peritoneal injection of Ketamine (75 mg/kg) + Xylazine (15 mg/kg) in saline solution. Briefly, stock suspensions of 2 mg/ml NP in 0.9% NaCl were vortexed and bath sonicated for 10 min at 37 kHz just before administration. Each mouse received a volume of 25 μl of stock solution (50 μg dose, or after a 10x dilution in 0.9% NaCl 5 μg dose). CeO2 NP used in the present study are from the same supplier and batch as that described in [54]. Shape (spherical) and size (22.4 ± 0.2 nm) were determined by transmission electron microscopy (TEM), crystallinity (cerianite) by X-ray diffractometer, and specific surface area was estimated by Brunauer Emmett Teller analysis (42 ± 0.5 m2/g). Moreover, zeta potential (9.5 ± 0.6 mV at pH 7) and hydrodynamic diameter (1480 nm) were determined by dynamic light scattering. No endotoxin content (determined by the Limulus Amebocyte Lysate test) could be detected. Mice were sacrificed 24 h, 1 or 4 weeks later. At that time, each mouse was anesthetized by intraperitoneal injection, with a cocktail of 3.33% buprenorphine, 32.03% zoletil, 4.2% xylazine and 60.43% physiological saline (0.9% saline) at 5 μL/g, and the lungs were harvested and collected for further analysis [54]. A broncho-alveolar lavage (BAL) was performed in a subset of mice as previously described [55]. Briefly, lungs were washed twice with 1 ml of 0.9% saline, and total alveolar cells were collected by a centrifugation at 400 g for 15 min at 4 °C. The resultant cellular pellet was suspended in physiological saline. Cellular viability was assessed as > 90% by trypan blue exclusion and the total number of cells were quantified. For differential counts, the cell suspension was spun (Cytospin-2, Shandon Products Ltd.), fixed in methanol, and stained using Diff Quick solution (Medion Diagnostics, Plaisir, France). Myeloid cell specific Atg5 deficient mice (Atg5fl/fl LysM-Cre+/− mice further referred as Atg5+/−) and their littermate wildtype (WT - Atgfl/fl LysM-Cre−/− − C57Bl/6 background) counterpart were kindly provided by Fatima Clerc [56]. GFP-LC3 mice (C57Bl/6 background) were purchased from Riken, Japan. All mice were subjected to the same procedures. The experimental protocol received the approval of the French Government (Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation, APAFIS #14914–2,018,042,515,599,016).
Histological and immunohistochemistry analyses
Paraffin embedded lung tissue sections (5 μm) were stained with Hematoxylin and Eosin or Sirius Red for histological observations and total collagen deposition, respectively. Immunohistochemistry experiments were performed using antibodies described in Table 2, after a pre-blocking step (incubation of tissue sections with 2.5% horse serum in PBS for 30 min). The duration of the primary antibody incubation was over night at 4 °C. We used biotinylated goat anti-rabbit secondary antibodies for Collagen I, Collagen III, SMA, TGF-ß, CD68, CD80, iNOS, CD163, CD206, Arginase1 (Vector Labs), biotinylated goat anti-rat secondary antibody for CD107b (Vector Labs), horse anti-rabbit IgG (Vector Labs) for Atg5, Alexa Fluor 488 (Green) for LC3 and Alexa Fluor 546 (Red) secondary antibodies for LAMP1 (Invitrogen).
At least 10 fields per lung tissue section (magnification 200X) were evaluated for the quantification of histological lesions and immunostainings using ImageJ software as previously described [54]. Briefly, for alveolar wall thickening, alveolar images without bronchi were used to build a macro with threshold value for alveolar wall thickness excluding alveolar spaces. For bronchiolar thickening, at least 10 fields with well defined, large and round shaped bronchi were imaged per animal. The thickness of each individual bronchus was measured manually at four different regions in pixels, and the average pixels calculated from these regions was converted into μm for bronchiolar thickening.
For immunohistological analyses, a representative image of alveolar or bronchiolar regions was opened under ImageJ and zoomed in for 3 colors. The plug-in with color deconvolution was applied on the image followed by choosing color 1 for DAB stain, color 2 for counter-stain, and color 3 for the white background. The next step was adjusting the threshold for 3 color regions and the specific threshold values for each color were noted and close-all option was employed. The image was then opened and plugin-macro-record and plugin-color deconvolution were applied to the image step-wise. Threshold was adjusted, and the noted values for color 2 and color 1 were set for analyzing the measure for each color.
Primary cells culture
Peritoneal macrophages: Briefly, primary cultures of peritoneal macrophages were obtained as previously described [57]. Briefly, 2 ml of sterile 4% thioglycolate broth (T0157, Sigma-Aldrich, La Verpillère, France) were administered in the peritoneal cavity of C57Bl/6 mice beforehand anesthetized by intraperitoneal injection using a cocktail of 3.33% buprenorphine, 32.03% zoletil, 4.2% xylazine and 60.43% physiological saline (0.9% saline) at 5 μL/g, using a 26G needle. Seventy-two hours later, mice underwent a cervical dislocation, and peritoneal macrophages were harvested from the peritoneal cavity and cultured in DMEM medium, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. For immunocytochemistry experiments, cells were seeded in 8-well cell culture chamber slides (LabTek, Nunc, ATGC Biotechnology, France) and exposed for 24 h to 10 μg/ml CeO2 NP. After incubation with the primary antibody of interest (Table 2), cells were labelled with secondary antibodies: Alexa Fluor 488 (green) for LC3 and Alexa Fluor 546 (red) for LAMP1. The fluorescence images were captured using Zeiss LSM-510 multitracking laser scanning confocal microscope with a Helium/Neon laser at 543 nm and using AxioVision software (Carl Zeiss).
Lung fibroblasts
Primary fibroblasts were isolated from C57Bl/6 mouse lungs by mechanic dissection and enzymatic digestion (collagenase 4,1% in HBSS). They were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and Glutamax (Gibco, 31,966–021) containing 10% foetal bovine serum (Eurobio) and streptomycin penicillin (Life Technologies, E1740384 100 μg/ml). Fibroblasts were seeded in 8-well Labtek for 24 h (10 000 cells per well) before being incubated for 48 h with the supernatant of CeO2 NP-exposed peritoneal macrophages obtained from WT or Atg5 animals. Immunofluorescence for the protein of interest was then performed as described for peritoneal macrophages, using Alexa Fluor 546 (red).
X-ray microfluorescence experiments
The localization and speciation of Ce in the lungs were assessed on lung tissue sections embedded in paraffin, using X-ray microfluorescence (micro-XRF) and micro X-ray absorption near edge structure (micro-XANES) respectively, as previously described [54]. Briefly, paraffin-embedded lung tissue sections (10 μm thick) were obtained from WT and Atg5+/− animals, exposed or not to CeO2 NP (50 μg, observation at 28 days). Sections were placed between two ultralene foils. These experiments were performed at the LUCIA beamline of the SOLEIL synchrotron (Orsay, France - [58, 59]), at 5.8 keV, just above the Ce L1 edge. The beam focalization was assured by a Kirkpatrick Baez (KB) mirror, which allows a beam size of 3.5*2.5 μm2 to be reached. The localization of CeO2 NP in lung tissue was visualized by map** the X-ray fluorescence of S. The very efficient flyscan mode was used. Micro-XANES spectra at the Ce L-edge were then collected in areas presenting high S concentrations (attesting of accumulation to lung tissue characteristic of fibrotic lesions). XANES data were obtained after performing standard procedures for pre-edge substraction and normalization using IFEFFIT implemented in the ATHENA® software package [60]. A total of 29 spectra in WT and 22 spectra in Atg5+/− animals have been recorded.
Statistical analysis
Five to eight mice per experimental group were utilized. Taking into account the possibility of non-normal distribution in the mice population, nonparametric tests (Kruskal–Wallis statistical test followed by Dunn’s multiple comparison test) were used [61]. Values are expressed as the mean ± SEM. Data were analyzed with GraphPad Prism 6.0 (La Jolla, CA) and STATA v13.0 (College Station, TX). For all statistical tests, p values smaller than 0.05 were considered as significant.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BAL:
-
Broncho-alveolar lavage
- CeO2 :
-
Cerium Oxide
- FBS:
-
Fetal Bovine Serum
- XANES:
-
X-ray absorption near edge structure
- XRF:
-
X-ray microfluorescence
- NP:
-
Nanoparticles
- REE:
-
Rare earth elements
References
Gwenzi W, Mangori L, Danha C, Chaukura N, Dunjana N, Sanganyado E. Sources, behaviour, and environmental and human health risks of high-technology rare earth elements as emerging contaminants. Sci Total Environ. 2018;636:299–313 [cited 2020 Sep 11]. Available from: https://doi.org/10.1016/j.scitotenv.2018.04.235.
Cassee FR, van Balen EC, Singh C, Green D, Muijser H, Weinstein J, et al. Exposure, health and ecological effects review of engineered nanoscale cerium and cerium oxide associated with its use as a fuel additive. Crit Rev Toxicol. 2011;41(3):213–29 Available from: https://www.ncbi.nlm.nih.gov/pubmed/21244219.
Pairon JC, Roos F, Iwatsubo Y, Janson X, Billon-Galland MA, Bignon J, et al. Lung retention of cerium in humans. Occup Environ Med. 1994;51:195–9.
Porru S, Placidi D, Quarta C, Sabbioni E, Pietra R, Fortaner S. The potencial role of rare earths in the pathogenesis of interstitial lung disease: a case report of movie projectionist as investigated by neutron activation analysis. J Trace Elem Med Biol. 2001;14:232–6.
Ma J, Bishoff B, Mercer RR, Barger M, Schwegler-Berry D, Castranova V. Role of epithelial-mesenchymal transition (EMT) and fibroblast function in cerium oxide nanoparticles-induced lung fibrosis. Toxicol Appl Pharmacol. 2017;323:16–25.
Ma JY, Mercer RR, Barger M, Schwegler-Berry D, Scabilloni J, Ma JK, et al. Induction of pulmonary fibrosis by cerium oxide nanoparticles. Toxicol Appl Pharmacol. 2012;262(3):255–64 Available from: https://www.ncbi.nlm.nih.gov/pubmed/22613087.
Ma JY, Young SH, Mercer RR, Barger M, Schwegler-Berry D, Ma JK, et al. Interactive effects of cerium oxide and diesel exhaust nanoparticles on inducing pulmonary fibrosis. Toxicol Appl Pharmacol. 2014;278(2):135–47 Available from: https://www.ncbi.nlm.nih.gov/pubmed/24793434.
Park E-J, Cho W-S, Jeong J, Yi J, Choi K, Kim Y, et al. Induction of inflammatory responses in mice treated with cerium oxide nanoparticles by Intratracheal instillation. J Heal Sci. 2010;56(4):387–96 Available from: http://joi.jlc.jst.go.jp/JST.JSTAGE/jhs/56.387?from=CrossRef.
Guo C, Robertson S, Weber RJM, Buckley A, Warren J, Hodgson A, et al. Pulmonary toxicity of inhaled nano-sized cerium oxide aerosols in Sprague–Dawley rats. Nanotoxicology. 2019;13(6):733–50.
Dikic I, Elazar Z, et al. Nat Rev Mol Cell Biol. 2018;19(6):349–64.
Nakahira K, Pabon Porras MA, Choi AMK. Autophagy in Pulmonary Diseases. Am J Respir Crit Care Med. 2016; Available from: https://doi.org/10.1164/rccm.201512-2468SO.
Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14(2):221–32 Available from: https://doi.org/10.1080/15548627.2017.1389823.
Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7(7):e41394 Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22815997.
Cohignac V, Landry MJ, Ridoux A, Pinault M, Annangi B, Gerdil A, et al. Carbon nanotubes, but not spherical nanoparticles, block autophagy by a shape-related targeting of lysosomes in murine macrophages. Autophagy. 2018;14(8):8.
Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in Fibrosing lung disease? Trends Mol Med. 2016;22(4):303–16 Available from: https://www.ncbi.nlm.nih.gov/pubmed/26979628.
O’Dwyer DN, Ashley SL, Moore BB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2016;311(3):L590–601.
Venosa A, Malaviya R, Gow AJ, Hall L, Laskin JD, Laskin DL. Protective role of spleen-derived macrophages in lung inflammation, injury, and fibrosis induced by nitrogen mustard. Am J Physiol Lung Cell Mol Physiol. 2015;309(12):L1487–98.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62 Available from: http://www.sciencedirect.com/science/article/pii/S107476131630053X.
Aalapati S, Ganapathy S, Manapuram S, Anumolu G, Prakya BM. Toxicity and bio-accumulation of inhaled cerium oxide nanoparticles in CD1 mice. Nanotoxicology. 2014;8(7):786–98.
Ma JY, Zhao H, Mercer RR, Barger M, Rao M, Meighan T, et al. Cerium oxide nanoparticle-induced pulmonary inflammation and alveolar macrophage functional change in rats. Nanotoxicology. 2011;5(3):312–25.
Zhu Y, Li H, Ding S, Wang Y. Autophagy inhibition promotes phagocytosis of macrophage and protects mice from methicillin-resistant Staphylococcus aureus pneumonia. J Cell Biochem. 2018;119(6):4808–14 Available from: http://doi.wiley.com/10.1002/jcb.26677.
Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019;26(4):715–27.
Bonilla DL, Bhattacharya A, Sha Y, Xu Y, **ang Q, Kan A, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39(3):537–47.
Hoppstädter J, Seif M, Dembek A, Cavelius C, Huwer H, Kraegeloh A. M2 polarization enhances silica nanoparticle uptake by macrophages. Front Pharmacol. 2015;6 Available from: https://doi.org/10.3389/fphar.2015.00055.
Auffan M, Rose J, Orsiere T, De Meo M, Thill A, Zeyons O, et al. CeO2 nanoparticles induce DNA damage towards human dermal fibroblasts in vitro. Nanotoxicology. 2009;3(2):161–71.
Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy. 2014;10(2):192–200.
Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93(6):875–81 Available from: http://www.jleukbio.org/content/93/6/875.abstract.
Zhang L, Wang Y, Wu G, **ong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res. 2018;19:170.
Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79:541–66.
Guo Y, Lin C, Xu P, Wu S, Fu X, **a W, et al. AGEs Induced Autophagy Impairs Cutaneous Wound Healing via Stimulating Macrophage Polarization to M1 in Diabetes. Sci Rep. 2016;6:36416.
Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11(2):271–84 Available from: https://www.ncbi.nlm.nih.gov/pubmed/25650776.
Zhao X, Wei S, Li Z, Lin C, Zhu Z, Sun D, et al. Autophagic flux blockage in alveolar epithelial cells is essential in silica nanoparticle-induced pulmonary fibrosis. Cell Death Dis. 2019;10(2) [cited 2020 Dec 21]; Available from: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/30755584/.
Mizumura K, Cloonan S, Choi ME, Hashimoto S, Nakahira K, Ryter SW, et al. Autophagy: friend or foe in lung disease? Ann Am Thorac Soc. 2016;13(Suppl 1):S40–7 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27027951.
Hill C, Li J, Liu D, Conforti F, Brereton CJ, Yao L, et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019;10(8):591 [cited 2020 Sep 3]. Available from: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/31391462/.
Jessop F, Hamilton RF, Rhoderick JF, Fletcher P, Holian A. Phagolysosome acidification is required for silica and engineered nanoparticle-induced lysosome membrane permeabilization and resultant NLRP3 inflammasome activity. 2017 [cited 2020 Sep 24]; Available from: https://doi.org/10.1016/j.taap.2017.01.012
Liu H, Cheng Y, Yang J, Wang W, Fang S, Zhang W, et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017;8(3):e2657.
Liu H, Fang S, Wang W, Cheng Y, Zhang Y, Liao H. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part Fibre Toxicol. 2016;13 Available from: https://doi.org/10.1186/s12989-016-0167-z.
Li Y, Liu R, Wu J, Li X. Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases [Internet], vol. 10: Theranostics. NLM (Medline); 2020. p. 7993–8017. [cited 2020 Sep 3]. Available from: /pmc/articles/PMC7381749/?report=abstract
Ghavami S, Yeganeh B, Zeki AA, Shojaei S, Kenyon NJ, Ott S, et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2018;314(3):L493–l504.
Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. 2016;309:101–10.
Cohignac V, Landry MJ, Boczkowski J, Lanone S. Autophagy as a possible underlying mechanism of nanomaterial toxicity. Nanomaterials. 2014;4:548–82.
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–63 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22286270.
Lee J, Kim HR, Quinley C, Kim J, Gonzalez-Navajas J, Xavier R, et al. Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem. 2012;287(6):4033–40 Available from: http://www.jbc.org.proxy.insermbiblio.inist.fr/content/287/6/4033.full.
Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–97 Available from: http://www.ncbi.nlm.nih.gov/pubmed/21228274.
Braga TT, Agudelo JSH, Camara NOS. Macrophages during the fibrotic process: M2 as friend and foe, vol. 6: Frontiers in Immunology. Frontiers Research Foundation; 2015.
Singh R, Kaushik S, Wang Y, **ang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–5 [cited 2021 Jan 7]. Available from: https://www-nature-com.proxy.insermbiblio.inist.fr/articles/nature07976.
Chen S, Yuan J, Yao S, ** Y, Chen G, Tian W, et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11(12):2346–57 [cited 2021 Jan 7]. Available from: https://www.tandfonline.com/doi/full/10.1080/15548627.2015.1109765.
Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. 2018;128(1):64–73.
Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–49.
Bachoual R, Boczkowski J, Goven D, Amara N, Tabet L, On D. Biological effects of particles from the paris subway system. Chem Res Toxicol. 2007;20 Available from: https://doi.org/10.1021/tx700093j.
Di YP, Tkach AV, Yanamala N, Stanley S, Gao S, Shurin MR, et al. Dual acute proinflammatory and antifibrotic pulmonary effects of short palate, lung, and nasal epithelium clone-1 after exposure to carbon nanotubes. Am J Respir Cell Mol Biol. 2013;49(5):759–67 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23721177.
Hevia H, Varela-Rey M, Corrales FJ, Berasain C, Martinez-Chantar ML, Latasa MU, et al. 5′-methylthioadenosine modulates the inflammatory response to endotoxin in mice and in rat hepatocytes. Hepatology. 2004;39(4):1088–98 Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15057913.
Tabet L, Bussy C, Setyan A, Simon-Deckers A, Rossi MJ, Boczkowski J, et al. Coating carbon nanotubes with a polystyrene-based polymer protects against pulmonary toxicity. Part Fibre Toxicol. 2011;8:3.
Paul E, Franco-Montoya M-L, Paineau E, Angeletti B, Vibhushan S, Ridoux A, et al. Pulmonary exposure to metallic nanomaterials during pregnancy irreversibly impairs lung development of the offspring. Nanotoxicology. 2017;11(4):484–95.
Presume M, Simon-Deckers A, Tomkiewicz-Raulet C, Le Grand B, Tran Van Nhieu J, Beaune G, et al. Exposure to metal oxide nanoparticles administered at occupationally relevant doses induces pulmonary effects in mice. Nanotoxicology. 2016;10:1–10.
Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11(8):1280–92 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26061908.
Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Vol. CHAPTER, Current Protocols in Immunology. NIH Public Access; 2008. p. Unit.
Vantelon D, Trcera N, Roy D, Moreno T, Mailly D, Guilet S, et al. The LUCIA beamline at SOLEIL. J Synchrotron Radiat. 2016;23:635–40.
Flank AM, Cauchon G, Lagarde P, Bac S, Janousch M, Wetter R, et al. LUCIA, a microfocus soft XAS beamline. Nucl Instruments Methods Phys Res Sect B Beam Interact Mater Atoms. 2006;246:269–74.
Ravel B, Newville M. ATHENA and ARTEMIS: Interactive graphical data analysis using IFEFFIT. Phys Scr T. 2005;115:1007–10.
Weissgerber TL, Milic NM, Winham SJ, Garovic VD. Beyond bar and line graphs: time for a new data presentation paradigm. PLoS Biol. 2015;13(4):e1002128. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25901488.
Acknowledgements
Authors wish to acknowledge the expertise of Erwan Paineau for the XRF analyses, and the expert advice of Fatima Clerc on Atg5+/- mice.
Funding
This work was supported by a funding of the French “Investissement d’Avenir” program LabEx SERENADE project (B. Annangi recipient of Research Chair).
Author information
Authors and Affiliations
Contributions
BA contributed to the design of experimental work, acquisition and analysis of data, and wrote the first draft of the manuscript. ZL, JBr, AR and VMS contributed to acquisition of data, DV contributed to acquisition and analysis of data, JBo critically revised the manuscript, and SL designed the work and critically wrote and revised the manuscript. The authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experimental protocol received the approval of the French Government (Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation, APAFIS #14914–2,018,042,515,599,016).
Consent for publication
Not applicable.
Competing interests
No competing interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Cell differential in CeO2-exposed mice. Quantification of cell differential in BAL fluid 24 h (Panel A), 1 week (panel B) or 28 days (Panel C) post-exposure to CeO2 NP (50 μg, C57Bl/6 mice). Each individual circle represents the value obtained from one animal (empty circle: saline exposure – plain circle: CeO2 NP-exposure). *p < 0.05.
Additional file 2: Figure S2.
Activation of autophagy in vitro. Panel A: Peritoneal macrophages of GPF-LC3 mice exposed to vehicle (Control) or 10 μg/ml CeO2 NP (CeO2). Panel B: Expression of Atg5 in peritoneal macrophages from C57Bl/6 CeO2-exposed mice. Blue color is for DAPI (nucleus) and green is for Atg5. Panel C: colocalization of LC3 (green) and LAMP1 (red) expression in GFP-LC3 mouse peritoneal macrophages in response to vehicle or CeO2 NP. Scale bar: 10 μm.
Additional file 3: Figure S3.
Expression of Atg5 peritoneal macrophages of CeO2-exposed mice. Expression of Atg5 in peritoneal macrophages from WT and Atg5+/− mice exposed to 10 μg/ml CeO2 NP for 6 h.
Additional file 4: Figure S4.
X-Ray microfluoresence and XANES spectra of CeO2-exposed mice. Representative images of XRF maps of Saline (Panel A and C) and CeO2 NP-exposed (Panel B and D) lungs from WT (Panel A and B) or Atg5+/− (Panel C and D) mice (observation at 28 days, 50 μg CeO2 NP). Original magnification × 100. False colors used in correlation XRF maps represent P (green), S (blue) and Ce (red). Panel E: XANES spectra at the Ce edge for Reference (blue line), and representative Ce spots (red, green and yellow lines).
Additional file 5: Figure S5.
Characterization of macrophage polarization in vitro. iNOS (Panel A), CD68 (Panel B), Arginase 1 (Panel C) or CD206 (Panel D) expression in mice peritoneal macrophages in response to Saline or 10 μg/ml CeO2 NP for 6 h.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Annangi, B., Lu, Z., Bruniaux, J. et al. Macrophage autophagy protects mice from cerium oxide nanoparticle-induced lung fibrosis. Part Fibre Toxicol 18, 6 (2021). https://doi.org/10.1186/s12989-021-00398-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-021-00398-y