
Abstract
Background
Free-range cattle are common in the Northeast China area, which have close contact with farmers and may carry virus threatening to cattle and farmers.
Methods
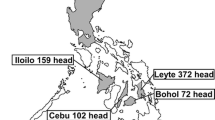
Using viral metagenomics we analyzed the virome in plasma samples collected from 80 cattle from the forested region of Northeast China.
Results
The virome of cattle plasma is composed of the viruses belonging to the families including Parvoviridae, Papillomaviridae, Picobirnaviridae, and divergent viral genomes showing sequence similarity to circular Rep-encoding single stranded (CRESS) DNA viruses. Five such CRESS-DNA genomes were full characterized, with Rep sequences related to circovirus and gemycircularvirus. Three bovine parvoviruses belonging to two different genera were also characterized.
Conclusion
The virome in plasma samples of cattle from the forested region of Northeast China was revealed, which further characterized the diversity of viruses in cattle plasma.
Similar content being viewed by others


Background
Although large-scale cattle farms account for an increasing part in the beef industry in China, free-range cattle raising is still used especially in the Northeast China area. Because free-range cattle have close contact with farmers, virus transmission from cattle to farmers through biting flies widely distributed in forested regions of Northeast China may conceivably occur. Using a metagenomics approach we characterized viral sequences present in pools of bovine serum samples collected from a forested region of Northeast China. Small circular, Rep-encoding, ssDNA (CRESS-DNA) genomes consist of a large and highly diverse collection of viruses [1,2,3] that infect a wide range of cellular hosts including vertebrates (Circoviridae), plants (Geminiviridae and Nanoviridae), crustaceans [4, 5], and fungi (SsHADV) [22]. The reads whose 5 bp to 55 bp from 5 prime end is identical were considered duplicates and only one random copy of duplicates was kept. Low sequencing quality tails were trimmed using Phred quality score ten as the threshold. Adaptors were trimmed using the default parameters of VecScreen which is NCBI BLASTn with specialized parameters designed for adapter removal. The cleaned reads were de-novo assembled within each barcode by SOAPdenovo2 version r240 using Kmer size 63 with default settings. The assembled contigs, along with singlets were aligned to an in-house viral proteome database using BLASTx with an E-value cutoff of <10−5. Candidate viral hits are then compared to an in-house non-virus non-redundant (NVNR) protein database, which was compiled using non-viral protein sequences extracted from NCBI nr fasta file, to remove false positive viral hits. Contigs without significant BLASTx similarity to viral proteome database are searched against viral protein families in vFam database [23] using HMMER3 [24,25,26] to detect remote viral protein similarities. A web-based graphical user interface was developed to present users with the virus hits, along with taxonomy information and processing meta-information.
Genome acquisition and PCR screening
To characterize the complete genome of the CRESS-DNA genomes, inverse PCR and Sanger sequencing was used with outward pointing PCR primers designed based on the initial Rep encoding contigs. Using the ORF finder of the Geneious software version 9, putative open reading frames (ORFs) and the stem-loop in the circular genomes were located. PCR screenings were also performed to investigate the prevalence of the five CRESS-DNA genomes in the 80 bovine plasma samples by nested PCR with specific primers designed based on the five genomes, respectively. Sequences and characteristics of the primers used in inverse and screening PCRs are shown in an additional file (see Additional file 1). PCR products were then subjected to Sanger sequencing.
Phylogenetic analysis
For generating a phylogenetic tree, sequence alignment was performed using CLUSTAL X with the default setting [27], phylogenetic analyses were constructed through full-length Rep protein of novel CRESS-DNA viruses and other genetically-close relatives. Using the Maximum-Likelihood method, phylogenetic trees with 1000 bootstrap resamples of the alignment data sets were generated, visualized using the program MEGA [28]. Bootstrap values (based on 1000 replicates) for each node are shown if >50%.
Result
Overview of eukaryotic viral sequences
The 80 cattle plasma samples in the five libraries generated a total of 2,493,791 unique sequence reads using the Illumina MiSeq sequencing run with 250 bases paired ends. Sequence reads were de novo assembled using the Ensemble program [29] and compared to the GenBank non-redundant protein database using BLASTx. Results indicated that CRESS-DNA sequences accounted for a major part of total putative mammalian virus reads, with 1848 reads showing sequence similarity to the viruses from Circoviridae family, and 2212 reads related to viruses from Genomoviridae family [30]. Table 1 list the other mammalian virus sequences detected namely bovine parvovirus (BPV) (2695 reads), papillomavirus (4 reads), and picobirnavirus (3 reads), where the sequence reads of papillomavirus and picobirnavirus were shown in an additional file (see Additional file 2). Besides these mammalian viruses, a small number of insect viral sequences were detected showing low-level sequence similarity to Iflaviridae and Dicistroviridae. The CRESS-DNA viruses showed lower levels similarities to known genomes available in GenBank, whose divergent genomes were then further characterized.
CRESS-DNA genomes
Two complete CRESS-DNA genomes showing the highest sequence similarity to viruses from Genomoviridae family were obtained through inverse PCR based on two large contigs from these libraries and Sanger sequencing. Genomes were 2192 nt (BGmv001, from library 5) and 2926 nt (BGmv002, from library 1) in length, respectively. As shown in Fig. 1a and b, the two genomes contained two bidirectional ORFs, encoding the putative Rep and Cap proteins. The two gemycircularviruses (the only genus currently in the Genomoviridae family) in this study did not contain putative intron in the Rep gene. BLASTp search in GenBank based on the amino acid sequence of Rep showed BGmv001 shared the highest sequence similarity of 56% to a gemycircularvirus genome sequenced from dragonfly abdomens (NC_023872) [8]. BGmv002 shared the highest protein sequence similarity of 36% to a gemycircularvirus from bird feces (KF371630) [11]. Figure 1 C showed the CRESS-DNA genome of Bvch001 with 2717 nt in length, whose Rep and Cap are arranged in inverse direction. The best matches in GenBank of Bvch001 based on amino acid sequence of Rep include two smacoviruses from Gorilla (KP233191 and KP233192) [31], two unidentified circular ssDNA virus from Rhesus macaque (KU043429 and KU043430) [32] and four porcine stool-associated circular viruses (KJ577812-KJ577815) [33], sharing 52% sequence similarity with them. Two complete CRESS-DNA genomes showing the highest similarity to circoviruses were determined through inverse PCR based on two large contigs from these libraries and Sanger sequencing, which were 1881 nt (Bvch002, from library 5) and 2926 nt (Bvch005, from library 4) in length, respectively. Figure 1 D and E indicated the genomic organization of Bvch002 and Bvch005, where the predicted Rep and Cap of Bvch002 are arranged in the same direction within the genome and those of Bvch005 are inversely arranged. BLASTp search in GenBank based on the amino acid sequence of Rep showed Bvch002 shared the highest sequence similarity of 38% to a circovirus-like virus from dragonfly (KM598397) and BGmv005 shared the highest sequence similarity of 48% to a bovine faeces-associated circular DNA virus (NC_030137) [34].

The genomic organization of the five CRESS-DNA viruses identified in the cattle blood samples
Based on the alignment of the Rep amino acid sequences herein detected with the best matches of BLASTp search in GenBank and those of representative CRESS-DNA genomes including circovirus, cyclovirus and gemycircularvirus, a phylogenetic tree was constructed. As shown in the Fig. 2 BGmv001 and BGmv002 belong to the cluster of gemycircularvirus, Bvch002 and Bvch005 fall into the clade showing close relationship with circovirus, and Bvch001 clustered into the group neighbouring to the gemycircularvirus clade.

Phylogenetic analysis of the five CRESS-DNA viruses identified in the cattle blood samples. Phylogenetic analysis was performed based on the amino acid sequence of Rep protein. The sequence alignments included five CRESS-DNA viruses identified in the cattle blood samples in this study, their best BLASTp matches in GenBank based on the Rep proteins, and the representative strains of circovirus, cyclovirus, and gemycircularvirus. Host or sources of the closely related viruses of the five CRESS-DNA viruses in this study in the phylogenetic analysis were showed on branches. Viruses identified in this study were labeled with black dots
PCR screening results indicated Bvch001 was detected in three samples, and sequencing results indicted the three 310 bp sequence fragments were different from each other, sharing >99.3% similarity (the two new sequences were deposited in GenBank with nos. MG601761 and MG601762). BGmv001 was detected in two samples and one of them (with GenBank no. of MG601763) had one base pair difference from the original genome based on the 290 bp PCR fragment. Two samples were positive for BGmv002 and the two sequences had two base pairs difference based on the 280 bp PCR product (the GenBank no. of new sequence is MG601764). The rest two CRESS-DNA genomes, Bvch002 and Bvch005, were respectively detected in a single sample, where their sequencing results were identical to the original genomic sequences, respectively.
Bovine parvovirus
Bovine parvovirus 3 (BPV3) was detected in all the five cattle plasma pools and BPV2 was detected in two pools. Three complete coding regions of BPV genomes could be assembled, where one (with stain name ujs2665) belongs to BPV2 (from CattleB03) and two (with stain name ujs1794 and ujs497) belong to BPV3 (from CattleB03 and CattleB04, respectively). Figure 3 showed the genomic organization of the three BPVs and phylogenetic trees based on the nonstructural (NS) and VP1 proteins, respectively. The BPV2’s NS showed 96–98% identities to the 3 BPV2 genomes currently available in GenBank database, while the VP protein showed 84–86% identities to them, suggesting that ujs2665 is a putative new strain in BPV2 group. The NS proteins of the two BPV3 shared 98% sequence identity with each other and both showed 96% sequence identity with the only BPV3 strains in GenBank, and the VP proteins of them shared 99% sequence identity with each other and 97% identity to the BPV strain.

The genomic organization of the three BPVs and phylogenetic trees based on the nonstructural (NS) and VP1 proteins, respectively. a The genomic organization of the three BPVs identified in the cattle blood samples. b and (c) Phylogenetic analysis based on the amino acid sequence of NS and VP proteins, respectively. The sequence alignments included three BPVs identified in the cattle blood samples in this study, their best BLASTp matches in GenBank, and the representative members of the related genera in family Parvoviridae. Viruses identified in this study were labeled with black dots
Nucleotide sequence accession numbers
The viral genomes described in detail here and the PCR screening sequence fragments were deposited in GenBank under the following accession numbers MG026727- MG026729, MF669476- MF669480, and MG601761- MG601764. The raw sequence reads from the viral metagenomic libraries were deposited in the Short Read Archive of GenBank database under the accession number SRX3235902.
Discussion
Our analysis characterizing enriched viral particle associated nucleic acids in cattle plasma showed that CRESS-DNA virus and bovine parvovirus sequences are present in all the five libraries and yielded the most reads relative to other viruses. In the recent years, a large number of CRESS-DNA genomes have been determined in mammals, birds, insects, plants, fungi, and environment samples which bringing to light a high level of genetic diversity among these virus [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. In this study, we detected three different groups of CRESS-DNA genomes in the plasma of free-range cattle in north-east China, which belonged to gemycircularvirus and two unclassified groups, respectively, based on phylogenetic analysis. Although detecting CRESS-DNA genomes in blood, a site considered to be sterile, makes it reasonable to believe the possibility of these viruses’ replication in the cell of cattle host, reports confirming replication in mammalian cells or of sero-conversion to these viruses are still lacking. It is also conceivably that CRESS-DNA genomes in blood results from skin contamination during phlebotomy, translocation from ingested food products or enteric organisms such as protozoa or fungi, or reflect systemic bovine infectious with still unknown cellular hosts with release of their viruses into the blood stream.
Bovine parvoviruses include BPV1, 2 and 3 and the recently described bosavirus belonging to three different genera of the Parvoviridae family. BPV most commonly causes diarrhoea in neonatal calves and respiratory and reproductive disease in adult cattle [35, 36]. In the present study, BPV3 was detected in all the five cattle plasma pools and BPV2 was detected in two pools, showing high viral sequence reads percentages. These cattle included in the present were all in normal status without clinical symptoms, thus detecting BPV3 with high rate in plasma samples from healthy cattle suggested this virus were prevalent in high rate in adult cattle in this area. BPV2 has also been reported in serum from calves in the US [37, 38].
The other four types of virus including papillomavirus, picobirnavirus, pegivirus, and bovine torovirus were also detected in cattle plasma pools with low percentage of sequence reads. The papillomavirus sequences detected in pool ID cattle02 were closely related to viruses detected in bovine teat papillomas [39]. The other pool ID cattle03 contained papillomavirus sequences showing <50% sequence similarity to human papillomavirus, which may represent a novel species of papillomavirus. Whether these papillomavirus sequences reflect contamination with human skin or bovine papillomaviruses possibly introduced into the serum pools from cattle skin during sampling is unknown. Picobirnavirus are widely present in animal fecal samples [40, 41], although they were detected in one plasma pool in the present study, they might be from the contamination of feces which is very common on bovine tail skin. Whether these viruses detecting in cattle plasma can cause disease needs further research.
In the investigation of virome, most research groups performed viral purification by filtering samples through small-pore (0.2–0.45 μm) filters, which prevented the detection of viruses larger than the filter pores, e.g. the members of the order Megavirales [42]. In our studies, considering that most members of giant viruses have diameter < 0.45 μm, we only used the 0.45 μm filters, which did not have significant effect on the virome results [21, 43].
Conclusion
The virome in plasma samples of cattle from the forested region of Northeast China included the viruses belonging to the families including Parvoviridae, Papillomaviridae, Picobirnaviridae, and viruses showing sequence similarity to CRESS DNA viruses, where 5 divergent genomes of CRESS-DNA viruses and 3 genomes of bovine parvoviruses were characterized in detail. This study increased the knowledge of the diversity of viruses in cattle plasma.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- BPV:
-
Bovine parvovirus
- CRESS:
-
Circular Rep-encoding single stranded
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
References
Delwart E, Li L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other rep encoding small circular ssDNA genomes. Virus Res. 2012;164:114–21.
Rosario K, Duffy S, Breitbart M. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch Virol. 2012;157:1851–71.
Zhang W, Li L, Deng X, Blümel J, Nübling CM, Hunfeld A, Baylis SA, Delwart E. Viral nucleic acids in human plasma pools. Transfusion. 2016;56:2248–55.
Dunlap DS, Ng TFF, Rosario K, Barbosa JG, Greco AM, Breitbart M, Hewson I. Molecular and microscopic evidence of viruses in marine copepods. Proc Natl Acad Sci. 2013;110:1375–80.
Ng T, Alavandi S, Varsani A, Burghart S, Breitbart M. Metagenomic identification of a nodavirus and a circular ssDNA virus in semi-purified viral nucleic acids from the hepatopancreas of healthy Farfantepenaeus Duorarum shrimp. Dis Aquat Org. 2013;105:237–42.
Yu X, Li B, Fu Y, Jiang D, Ghabrial SA, Li G, Peng Y, **e J, Cheng J, Huang J, Yi X. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci U S A. 2010;107:8387–92.
Zawar-Reza P, Argüello-Astorga GR, Kraberger S, Julian L, Stainton D, Broady PA, Varsani A. Diverse small circular single-stranded DNA viruses identified in a freshwater pond on the McMurdo ice shelf (Antarctica). Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2014;26:132–8.
Rosario K, Dayaram A, Marinov M, Ware J, Kraberger S, Stainton D, Breitbart M, Varsani A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). The Journal of general virology. 2012;93(Pt 12):2668–81.
Dayaram A, Potter KA, Moline AB, Rosenstein DD, Marinov M, Thomas JE, Breitbart M, Rosario K, Argüello-Astorga GR, Varsani A. High global diversity of cycloviruses amongst dragonflies. The Journal of general virology. 2013;94(Pt 8):1827–40.
Rosario K, Seah YM, Marr C, Varsani A, Kraberger S, Stainton D, Moriones E, Polston JE, Duffy S, Breitbart M. Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and Old Worlds. Viruses. 2015;7(10):5553–70.
Sikorski A, Massaro M, Kraberger S, Young LM, Smalley D, Martin DP, Varsani A. Novel myco-like DNA viruses discovered in the faecal matter of various animals. Virus Res. 2013;177:209–16.
Smits SL, Zijlstra EE, van Hellemond JJ, Schapendonk CME, Bodewes R, Schürch AC, Haagmans BL, Osterhaus ADME: Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010–2011. Emerging infectious diseases. 2013;19:1511–13.
Phan TG, Kapusinszky B, Wang C, Rose RK, Lipton HL, Delwart EL. The fecal viral flora of wild rodents. PLoS Pathog. 2011;7:e1002218.
Shan T, Li L, Simmonds P, Wang C, Moeser A, Delwart E. The fecal virome of pigs on a high-density farm. J Virol. 2011;85:11697–708.
Reuter G, Boros Á, Delwart E, Pankovics P. Novel circular single-stranded DNA virus from turkey faeces. Arch Virol. 2014;159:2161–4.
Cheung AK, Ng TF, Lager KM, Bayles DO, Alt DP, Delwart EL, Pogranichniy RM, Kehrli ME. A divergent clade of circular single-stranded DNA viruses from pig feces. Arch Virol. 2013;158:2157–62.
Tijssen P, Pénzes JJ, Yu Q, Pham HT, Bergoin M. Diversity of small, single-stranded DNA viruses of invertebrates and their chaotic evolutionary past. J Invertebr Pathol. 2016;140:83–96.
Breitbart M, Delwart E, Rosario K, Segalés J, Varsani A, Ictv Report Consortium: ICTV Virus Taxonomy Profile: Circoviridae. The Journal of general virology. 2017;98(8):1997–8.
Li L, Deng X, Mee ET, Collot-Teixeira S, Anderson R, Schepelmann S, Minor PD, Delwart E. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J Virol Methods. 2014;213C:139–46.
Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL:An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Research 2015.
Zhang W, Yang S, Shan T, Hou R, Liu Z, Li W, Guo L, Wang Y, Chen P, Wang X, Feng F, Wang H, Chen C, Shen Q, Zhou C, Hua X, Cui L, Deng X, Zhang Z, Qi D, Delwart E. Virome comparisons in wild-diseased and healthy captive giant pandas. Microbiome. 2017;5:90.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Skewes-Cox P, Sharpton TJ, Pollard KS, DeRisi JL. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS One. 2014;9:e105067.
Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC bioinformatics. 2010;11:431.
Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome informatics International Conference on Genome Informatics. 2009;23:205–11.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG, Clustal W, Clustal X. Version 2.0. Bioinformatics (Oxford, England). 2007;23:2947–8.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015;43:e46.
Krupovic M, Ghabrial SA, Jiang D, Varsani A. Genomoviridae: a new family of widespread single-stranded DNA viruses. Arch Virol. 2016;161:2633–43.
TFF N, Zhang W, Sachsenröder J, Kondov NO, da Costa AC, Vega E, Holtz LR, Wu G, Wang D, Stine CO, Antonio M, Mulvaney US, Muench MO, Deng X, Ambert-Balay K, Pothier P, Vinjé J, Delwart E. A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus Evolution. 2015;1:vev017.
Kapusinszky B, Ardeshir A, Mulvaney U, Deng X, Delwart E. Case control comparison of enteric viromes in captive rhesus macaques with acute or idiopathic chronic diarrhea. J Virol. 2017;91(18):JVI.00952–17.
Cheung AK, Ng TFF, Lager KM, Alt DP, Delwart E, Pogranichniy RM. Identification of several clades of novel single-stranded circular DNA viruses with conserved stem-loop structures in pig feces. Arch Virol. 2015;160:353–8.
Steel O, Kraberger S, Sikorski A, Young LM, Catchpole RJ, Stevens AJ, Ladley JJ, Coray DS, Stainton D, Dayaram A, Julian L, van Bysterveldt K, Varsani A. Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect Genet Evol. 2016;43:151–64.
Ng TFF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E, Metagenomics A. Case-control study to identify viruses associated with bovine respiratory disease. J Virol. 2015;89:5340–9.
Woode GN, Reed DE, Runnels PL, Herrig MA, Hill HT. Studies with an unclassified virus isolated from diarrheic calves. Vet Microbiol. 1982;7:221–40.
Sadeghi M, Kapusinszky B, Yugo DM, Phan TG, Deng X, Kanevsky I, Opriessnig T, Woolums AR, Hurley DJ, Meng X-J, Delwart E. Virome of US bovine calf serum. Biologicals. 2017;46:64–7.
Toohey-Kurth K, Sibley SD, Goldberg TL. Metagenomic assessment of adventitious viruses in commercial bovine sera. Biologicals. 2017;47:64–8.
Ogawa T, Tomita Y, Okada M, Shinozaki K, Kubonoya H, Kaiho I, Shirasawa H. Broad-spectrum detection of papillomaviruses in bovine teat papillomas and healthy teat skin. J Gen Virol. 2004;85:2191–7.
Ganesh B, Bányai K, Martella V, Jakab F, Masachessi G, Kobayashi N. Picobirnavirus infections: viral persistence and zoonotic potential. Rev Med Virol. 2012;22:245–56.
Ganesh B, Masachessi G, Mladenova Z. Animal picobirnavirus. Virusdisease. 2014;25:223–38.
Colson P, Fancello L, Gimenez G, Armougom F, Desnues C, Fournous G, Yoosuf N, Million M, La Scola B, Raoult D. Evidence of the megavirome in humans. J Clin Virol. 2013;57:191–200.
Zhang W, Li L, Deng X, Kapusinszky B, Pesavento PA, Delwart E. Faecal virome of cats in an animal shelter. The Journal of general virology. 2014;95(Pt 11):2553–64.
Acknowledgements
Not applicable.
Funding
This work was partly supported by National Key Research and Development Programs of China No. 2017YFC1200201, National Natural Science Foundation of China No. 81741062 and 31572525, Jiangsu Provincial Key Research and Development Projects No. BE2017693, Projects of the State Forestry Administration No.2130211, and Blood Systems Research Institute.
Availability of data and materials
The raw sequence reads from the viral metagenomic libraries were deposited in the Short Read Archive of GenBank database under the accession number SRX3235902.
Author information
Authors and Affiliations
Contributions
WZ conceived the study. HW performed most of the experiments. WZ wrote the paper. All authors participated the experiments and read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Primers used in the specific screening PCR and inverse PCR. (PDF 54 kb)
Additional file 2:
The sequence reads of papillomavirus and picobirnavirus in library cattle02. (PDF 33 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, H., Li, S., Mahmood, A. et al. Plasma virome of cattle from forest region revealed diverse small circular ssDNA viral genomes. Virol J 15, 11 (2018). https://doi.org/10.1186/s12985-018-0923-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-0923-9