
Abstract
Multiple sclerosis (MS) is a chronic debilitating disease of the central nervous system primarily mediated by T lymphocytes with specificity to neuronal antigens in genetically susceptible individuals. On the other hand, myasthenia gravis (MG) primarily involves destruction of the neuromuscular junction by antibodies specific to the acetylcholine receptor. Both autoimmune diseases are thought to result from loss of self-tolerance, which allows for the development and function of autoreactive lymphocytes. Although the mechanisms underlying compromised self-tolerance in these and other autoimmune diseases have not been fully elucidated, one possibility is numerical, functional, and/or migratory deficits in T regulatory cells (Tregs). Tregs are thought to play a critical role in the maintenance of peripheral immune tolerance. It is believed that Tregs function by suppressing the effector CD4+ T cell subsets that mediate autoimmune responses. Dysregulation of suppressive and migratory markers on Tregs have been linked to the pathogenesis of both MS and MG. For example, genetic abnormalities have been found in Treg suppressive markers CTLA-4 and CD25, while others have shown a decreased expression of FoxP3 and IL-10. Furthermore, elevated levels of pro-inflammatory cytokines such as IL-6, IL-17, and IFN-γ secreted by T effectors have been noted in MS and MG patients. This review provides several strategies of treatment which have been shown to be effective or are proposed as potential therapies to restore the function of various Treg subsets including Tr1, iTr35, nTregs, and iTregs. Strategies focusing on enhancing the Treg function find importance in cytokines TGF-β, IDO, interleukins 10, 27, and 35, and ligands Jagged-1 and OX40L. Likewise, strategies which affect Treg migration involve chemokines CCL17 and CXCL11. In pre-clinical animal models of experimental autoimmune encephalomyelitis (EAE) and experimental autoimmune myasthenia gravis (EAMG), several strategies have been shown to ameliorate the disease and thus appear promising for treating patients with MS or MG.
Similar content being viewed by others


Background
Multiple sclerosis (MS) and myasthenia gravis (MG) are autoimmune diseases affecting the central nervous system (CNS) and the neuromuscular junction (NMJ), respectively. These diseases are characterized by inflammation, immune dysregulation, and immune over activity [1, 2]. Defects in self-tolerance leading to autoimmunity distinguish MS from other well-known neuroinflammatory diseases including Parkinson’s disease, Alzheimer’s disease, and stroke episodes [3]. Likewise, the autoimmune component of MG distinguishes it from many other muscular dystrophies [2]. MS and MG affect 50/100,000 and 20/100,000 people, respectively, in the USA; this prevalence amounts to more than the collective prevalence of other neurologically relevant autoimmune diseases including optic neuritis, Guillain-Barre Syndrome (GBS), and acute disseminated encephalomyelitis [4,5,6,7,8]. Current treatments for MS and MG involve non-specific mechanisms of action, do not produce lifelong protection from flare-ups, and have undesirable side effects including fatalities [9,10,11,12]. Since T regulatory cells (Tregs) characterized as CD4+CD25+FoxP3+ (forkhead box P3) cells have emerged pivotal in suppressing autoimmune diseases like type 1 diabetes and others, this review is focused on evaluating the role of Treg defects or dysfunctions in MS- and MG-related autoimmune pathology [1, 13–17]. It also discusses the beneficial effects of Treg augmentation as a potential treatment strategy. It does so by summarizing recent data from MS and MG patients, as well as their pre-clinical models: experimental autoimmune encephalomyelitis (EAE) and experimental autoimmune myasthenia gravis (EAMG), respectively.
Although MS and MG are distinct autoimmune diseases, some studies suggest a co-morbidity [18]. A retrospective analysis indicated that five out of 1718 patients with MS also had MG (0.29%; expected percentage (0.005–0.015%)) [18]. Notably, the severity of both MS and MG was mild and the MG symptoms were ocular with none of these patients having positive anti-acetylcholine receptor (AChR) antibodies [18]. In another study, MS preceded MG in five out of eight patients by 6 to 8 years [19]. Onset of MG in MS patients treated with interferon (IFN)-β and glatiramer acetate (GA) was reported in one [20], but not in another study [18]. Therefore, definitive evidence of co-morbidity between MS and MG requires further validation in large independent cohorts of patients.
Despite differences in pathologies of MS and MG, there are immunological similarities. Both MS and MG are considered largely T cell mediated [1, 21]. MS and MG patients have increased numbers of Th1 and Th17 cells along with their associated cytokines IL-1, IL-6, IL-17, IFN-γ, and tumor necrosis factor (TNF)-α (Table 1) [21,22,23,24,25,26]. Further, Tregs of these patients have numerous documented dysfunctionalities [1, 13, 27, 28]. T cells originate from the thymus, and evidence suggests the thymus as a key regulator in the pathogenesis MG, and less so in MS [18, 20, 29]. Thus, thymectomy is a treatment option from which few MS patients may benefit [30, 31]. Both MS and MG have altered recent thymic emigrants suggestive of lower thymic output (Table 1) [28, 29, 32]. Further, MG and MS thymuses have been found to contain clonally expanded B cell lines (Table 1), a characteristic not found in control patients, or patients with systemic lupus erythematosus (SLE), anther autoimmune disease [33]. Germinal centers appear in the pathogenesis of both diseases [34]. CXCR5, a receptor associated with germinal center (GC) migration, can be found expressed on both T and B cells, but is elevated on CD4+ cells in both diseases (Table 1) which correlates well with the disease severity [34]. Both MS and MG patients have evidence of antigen-specific T cells [35, 36]. While MG patients have definitive evidence of antigen-specific antibody-mediated damage to the NMJ, the presence of antigen-specific antibodies in MS is debatable [37,38,39]. Immunological similarities and differences become more evident with treatments using immune modulating agents. Rituximab, a B cell depleting anti-CD20 antibody, has proven beneficial in both diseases, indicating B cell importance for each [40, 41]. In contrast, MS and MG patients differ in their response to IFN-1 (i.e., alpha and beta) treatments (Table 1). Development of MG after IFN-α treatment has been previously reported, while treatment of MS with IFN-β has been shown to be beneficial [12, 42]. IFN-1 (IFN-β) has also led to increased autoantibody production, a possible reason for MG exacerbation upon IFN-1 (IFN-α) treatment [43]. Similarly, a patient with MS treated with GA was seen to develop MG [44]. Lastly, anti-TNF-α therapy has resulted in different effects in patients with MS and MG. In MS, anti-TNF-α therapy has been unsuccessful despite success in pre-clinical models [45]. Whereas a majority of MG patients receive benefit, although some receive no benefit or even disease exacerbation [45, 46]. These studies clearly indicate that the immunological perturbation in these two diseases may have some common features, but are dissimilar.
Autoimmune development may not only be influenced by inadequate Treg numbers or defective Treg function, but it is also influenced by effector T cells (Teff; CD4+FoxP3−) resistant to suppression [47]. Although this review focuses on restoring Treg numbers and deficits, Teff resistance should be briefly discussed. The local cytokine milieu of IL-2, IL-4, IL-6, IL-15, and TNF-α have all been shown to influence Teff resistance to suppression [48, 49]. In MS, a decrease in the frequency of Tregs and resistance of Teffs to suppression were noted [50,51,52]. Similarly, both Tregs and Teffs from MG patients were found to be defective in ex vivo studies [53]. Whereas FoxP3 inhibited Th17 differentiation via repression of transcription factor RORγt, exogenous provision of IL-6 supported the differentiation of Th17 cells, suggesting the plasticity of the T cell under appropriate conditions [54]. Genetic studies unraveled polymorphisms associated with molecules related to Treg function in MS and MG patients [55, 56]. Although these data suggest an intrinsic functional defect in Tregs (Table 1), it is not clear whether it is sufficient to impair the functionality of Tregs. However, the conversion of FoxP3+ Tregs derived from normal humans into Th17 cells under the influence of IL-1 and IL-2 ex vivo has been documented, supporting the plasticity of Tregs [57], also observed in mice [54]. This is also suggested from an experiment in EAMG noting that the Treg defects appear after disease induction but the disease itself can be suppressed upon adoptive transfer of ex vivo generated Tregs [58, 59]. Inasmuch as the Tregs appear to be defective in both MS and MG (Table 1), we have focused this review on both intrinsic and extrinsic factors affecting Treg function in these diseases [1, 13, 27, 28].
Main text
Implications of dysregulated Tregs in MS and MG
Tregs play a key role in maintaining self-tolerance, and their dysfunction is well documented in multiple autoimmune diseases including Type 1 diabetes, GBS, psoriasis, and others [1, 13–17]. Tregs regulate immune response in the periphery predominantly by suppressing Teff cells. Although significant differences in the number of circulating Tregs in MS or MG patients relative to healthy controls are not frequently reported, Tregs from these patients are reported to have lower suppressive capabilities [1, 13, 60, 61]. This suggests that functional deficits in Tregs may contribute to the pathogenesis of MS and MG. For example, defects in Treg suppressor molecules have been linked to MS, such as reduced IL-10 production and genetic variations in CD25 [27, 55]. Likewise, MG patients have documented dysregulation in cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) expression, IL-2 sensitivity, and the levels of transforming growth factor beta (TGF-β) gene expression [25, 62, 63]. Mechanistically, lower Treg suppressive capabilities may lead to enhanced production of pro-inflammatory cytokines such as IL-6, IL-17, and IFN-γ, as well as activation of autoantibody producing B cells. Normally, Tregs suppress the production of these pro-inflammatory cytokines through contact-dependent and contact-independent suppression of Teff cells known to produce IL-6, IL-17, and IFN-γ [64, 65]. Functional Tregs are also believed to directly eliminate both antigen-presenting and autoantibody-producing B cells in a contact-dependent manner through secretion of perforin and granzyme as seen in lupus prone mice (NZB/W) [66]. This mechanism may be pivotal for Treg-based amelioration of MG and perhaps MS [2, 66, 67]. The autoreactive B cells can produce autoantibodies against acetylcholine receptor (AChR) or muscarinic receptor, thus causing membrane damage via activation of the complement cascade in MG; likewise, it has been suggested that autoantibodies may also be involved in the pathogenesis of MS [68, 69]. Furthermore, activated B cells are found to be upregulated in the neuroinflammatory sites of the CNS in MS patients and in the blood of MG patients [55]. While CD127 is expressed highly on Teff, but minimally on Tregs, molecules such as GITR, OX40, Helios, CD49b are also expressed on Tregs [22, 28, 47, 77]. Furthermore, many MS patients have decreased FoxP3 expression in Tregs, decreased Treg suppressive function, and decreased levels of Tr1 [1, 27]. Additionally, recent research suggests that Tregs are unable to properly infiltrate the CNS during the course of the disease [104,105,106]. Brain biopsies from MS patients revealed that 30% of the lesions lacked FoxP3 expression [104]. Fas, a cellular apoptotic pathway receptor, is seen upregulated on Tregs in MS brain biopsies suggesting increased susceptibility to apoptosis [104]. Taken together, these findings suggest that Tregs might be restricted from migrating to neuroinflammatory sites or undergo apoptosis upon arrival. Treg dysfunction during the disease progression can also be noted through modulation of CD28 and CTLA-4 co-receptors mentioned earlier. Studies in EAE have shown that during relapse, there is increased expression of CD28, co-localizing with T cell receptor subunit CD3, in the CNS blood vessels and parenchyma [107]. During remission, CD28+ cells decrease while CTLA-4+ cells increase [107]. Considering Tregs constitutively express CTLA-4 while Teff do not, it is possible these cells are Tregs; however, the study did not look for co-expression of FoxP3 [107, 108]. Taken together, Treg abnormalities in MS appear to involve loss of suppressive capacity as well as defect in migration into the CNS.
Many treatments which increase Tregs in EAE have been found to be efficacious, while other treatments, although successful, showed no effect on Tregs [109]. Administration of an indoleamine 2,3-dioxygenase (IDO) metabolite which increases Treg number leads to significant amelioration of MOG-induced EAE [110]. Additionally, the effect of IL-10-based therapy has been evaluated in different EAE models; most resulted in reduced clinical scores, perhaps associated with an increase in Tr1 associated with IL-10 administration [111, 112]. Some of the most affirmative evidence advocating Treg augmenting treatments have utilized adoptive transfer techniques. Adoptive transfer of FoxP3− Treg cells that ectopically expressed the transcription factor FoxA1 (suggested marker for a novel Treg) ameliorated EAE in IFN-β knockout (KO) mice and these cells were increased in response to treatment of MS patients with IFN-β, suggesting a role for this Treg subset [113]. Although adoptive transfer of CD4+CD25+FoxP3+ cells reduced EAE, they differed in their efficacy. Adoptive transfer of CNS-derived KLRG1− and KLRG1+ cells modestly reduced the clinical score at the termination of the experiment without affecting peak response (KLRG1 is associated with natural killer cells but was tested due to its increased expression on Tregs when entering the CNS of EAE mice) [114]. Likewise, CNS-derived CD4+CD25+ cells reduced the peak clinical EAE score [115]. Finally, experiments knocking out known Treg proliferating molecules, such as IDO or IL-10, resulted in exacerbation of EAE [110, 116].
In contrast to the above findings, FoxP3gfp knock-in mice had an accumulation of Treg cells in the CNS, but it failed to control EAE [117]. Administration of cholesterol-reducing statins such as atorvastatin inhibited the development of EAE without increasing the frequency of FoxP3+ Treg cells or Th2 cells, yet there was a substantial increase in IL-10 expression [109]. Taken together, these data tend to suggest that modulation of FoxP3 Tregs may not always be necessary for the protective effect of certain treatment modalities for EAE. Inasmuch as administration of statins involve non-specific immunosuppression, it is hard to avoid undesirable consequences. Therefore, it appears that boosting the Treg cells may produce a desirable clinical outcome in MS.
Treg migratory receptor dysregulation may have implications in MS
Dysfunctional migration of Tregs to neuroinflammatory sites could have profound implications; for example, GCN2 (aids in CCL2-mediated migration) KO Tregs adoptively transferred into EAE mice were unable to enter the CNS and hence mice were unable to undergo remission [118]. Some chemoattractants (e.g., CCL17) or their receptors (e.g., CCR4 and CXCR3) appear to facilitate Treg homing to neuroinflammatory sites. Human CD4+CD25+ Treg cells express CCR4 which respond to mature dendritic cell-derived CCL17 and are thereby recruited to the site of inflammation. Butti et al. determined that CCR4 is indispensable for Treg recruitment into the cerebral spinal fluid (CSF) and that recruitment was necessary for EAE amelioration likely due to the increased CCL17 levels after administration of IL-4 gene therapy into the CSF [119]. Studies of malignant diseases also find a correlation between the levels of CCL17 and accumulation of FoxP3+ cells, implicating CCL17 as a Treg recruiter molecule [120, 121]. Therefore, CCL17 administration at specific sites may be utilized to recruit Tregs.
Molecules such as CXCR3 are important for Tr1 and Treg migration to the CNS [106, 122]. The chemokine receptor CXCR3 that promotes trafficking of activated T and natural killer cells in response to CXCL9, CXCL10, and CXCL11 is expressed heavily on Tr1 cells which can direct Tr1 to the CNS thereby implying a role in EAE [122]. Administration of CXCL10-Fc exacerbates EAE, whereas CXCL11-Fc leads to amelioration with subsequent polarization to Tr1 [122]. Since CXCR3 and CXCL10 are found elevated in neuroinflammatory sites in MS patients, over-recruitment of Teff may be occurring, thus use of CXCL11 could be a strategy to recruit additional Tr1 to these neuroinflammatory sites [123]. Additionally, CXCR3 KO mice have more severe EAE with reduced FoxP3+ cells in the CNS despite little change in total CD4+ infiltrates implying that CXCR3 may be crucial for Treg migration to the CNS [106]. Therefore, a strategy to increase CXCR3+ expression on FoxP3+ Tregs may be useful for recruitment to the CNS. Enhanced CXCR3 expression by Tregs has been seen with the use of IL-27, thus providing a treatment strategy to facilitate selective Treg migration [122, 124].
Therapeutics for multiple sclerosis which restore Treg abnormalities
Many common MS treatments could result in adverse effects revealing a need for new therapeutic strategies [9, 125]. Additionally, current treatments such as IFN-β (Avonex, Rebif, and Betaseron), glatiramer acetate (GA; Copaxone), fingolimod (Gilenya), teriflunomide (Aubagio), dimethyl fumarate (Tecfidera), Natalizumab (Tysabri), and mitoxantrone, although beneficial, are not curative [9]. Corticosteroids, which have been in use for over 75 years, are still the treatment of choice for acute MS symptoms [9, 126]. However, they lack specificity and tend to cause severe adverse effects such as abnormal weight gain, behavioral changes, oral thrush, and others [126]. Over two decades ago, glatiramer acetate became one of the first treatments for reducing relapse rates in MS patients; however, it had only a modest efficacy in reducing disease activity [127]. Many newer treatments, such as the ones indicated above, have been implemented since then, yet nearly all can cause significant side effects [9, 128, 129]. Therefore, there is a need for novel treatments which reduce disadvantages of current treatments while providing greater efficacy.
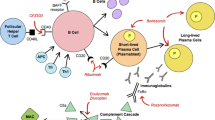
Most of the clinical treatments do not target Tregs as specifically as new pre-clinical treatments can [130]. Despite different targets, some treatments have been shown to increase Treg numbers such as IFN-β, GA, fingolimod, and teriflunomide, while others, such as dimethyl fumarate, natalizumab, mitoxantrone, and corticosteroids, have resulted in reduced, unaffected, or contradictive effects on Treg numbers [131,132,133,134,135,136,137,138,139,140]. Treatments such as the lipid-lowering drug atorvastatin mentioned earlier have no effect on the FoxP3 Treg population, indicating that increasing the Treg population, although may be desirable, may not always be necessary [109]. However, this review will discuss strategies to restore Treg abnormalities as therapeutic modalities that may be considered for further testing. These Treg abnormalities have been corrected using cytokines such as IL-10, IL-27, IL-35, bimolecular protein inhibitors (BPIs), indoleamine 2,3-dioxygenase (IDO), or the chemokine CXCL11 (Table 2; Fig. 1) [80, 106, 110, 111, 141]. Additionally, we propose modalities of treatment for further testing. These include Jagged-1/OX40L co-treatment and site-specific CCL17 administration for Treg recruitment (Table 2).

MG and MS treatment schemes aimed at augmenting Tregs based on experimental models. AChR: acetylcholine receptor; APC: antigen-presenting cell; B7AP: B7 antisense peptide; Bimolecular peptide inhibitor; CNS: central nervous system; DC: dendritic cell; GM-CSF: granulocyte macrophage-colony stimulating factor; IDO: indoleamine 2,3-dioxygenase; iTr35: induced T-regulatory 35 cell; IFN-γ: interferon gamma; MG: myasthenia gravis; MS: multiple sclerosis; N3: Notch 3 receptor; IL: interleukin; Teff: effector T cell; TFH: follicular helper T cell; TFR: regulatory T follicular cell; TGF-β: transforming growth factor beta: Tr1: T-regulatory 1 cell; Treg: T-regulatory cell
Administration of interleukins
IL-10 mediated suppression is regarded as a main mechanism of Treg suppression. IL-10 therapies have resulted in a reduction of symptoms in majority of the EAE models tested (reviewed in reference [111]) [142, 143]. Despite the success in pre-clinical models, it has not been as successful in clinical trials conducted in other autoimmune diseases such as Crohn’s disease and rheumatoid arthritis [144, 145]. Focus might instead be diverted toward IL-10 replenishing strategies which down modulates the Fas-mediated apoptosis both in Tregs and Bregs [111, 145]. Research shows increasing IL-10 expression may be done through upregulating IL-10 producing Tr1, increasing Treg number, or through induction of tolerogenic DCs (CD11c+CD8α−) [27, 67, 110]. Since many documented positive correlations between IL-10 and MS remission have been documented, and IL-10 secreting Tr1 usage in pre-clinical models appears useful, upregulation of Tr1 cells could be a useful IL-10 replenishment strategy which would also rectify Tr1 defects in MS [146, 147].
IL-27 and IL-35 are anti-inflammatory cytokines that can regulate Treg responses [148, 149]. IL-35 has been shown to play a key role in FoxP3+ Treg suppression and in the induction of iTr35 [80, 149]. Treatment with IL-35 has shown to lead to EAE amelioration with concomitant induction of iTr35 (Fig. 1) [80]. These iTr35 have also been shown to ameliorate other autoimmune models such as collagen-induced arthritis and IBD [150]. IL-35 is not produced by human FoxP3 Tregs, but ex vivo experiments on human T cells have been able to induce iTr35 suggesting that it may have a similar role in humans [150, 151]. IL-27 has both inflammatory and anti-inflammatory pathways. IL-27 activates Th1 response through T-bet signaling; however, it also inhibits Th17 development and induces IL-10 production [152]. IL-27 role in FoxP3 Tregs is less understood, yet it has been shown to induce Tr1 production in vivo leading to EAE amelioration [153]. Administration of exogenous IL-27 to EAE mice has also been shown to reduce IL-17 production, Th17 cells, and CNS infiltration [153, 11]. Although the mechanism is not understood, Tregs have been seen to modestly increase with the use of drugs not targeting Tregs, such as pyridostigmine, rituximab, azathioprine, and IVIG [190,191,192,193]. Corticosteroids lack specificity and importantly cause severe adverse effects such as bruising, abnormal weight gain, behavior changes, oral thrush, and others [194]. Pyridostigmine bromide, the most commonly used acetylcholinesterase inhibitor, is the first line for symptomatic treatment [195]. Yet it provides only short-term relief, it is not a disease-modifying therapeutic (DMT), and it can cause a hypersensitivity rash [195]. Azathioprine, a commonly used DMT, causes side effects in roughly 20% of patients and failed to prevent relapse in roughly 33% of patients in a long-term study of 117 MG patients [196]. Thymectomy has proven to be a beneficial DMT; however, it does not cure the underlying disease, and patients still suffer side effects from other treatments because thymectomized patients still require additional interventions such as corticosteroids or azathioprine [197]. Taken together, there is a need for more efficacious DMTs focused on destroying autoreactive B cells to reduce autoantibody production and NMJ destruction. Although anti-B cell therapies are in practice like rituximab (anti-CD20 antibody), Tregs’ ability to destroy autoreactive B cells may provide relatively better specificity [68, 92]. Treg-augmenting therapies which might accomplish this task or treat MG Treg abnormalities include GM-CSF, IL-2/mAb complexes, OX40L-Jagged-1 co-treatment, and TGF-β administration.
GM-CSF
GM-CSF treatment of MG has led to a reduction of clinical signs and symptoms while increasing Tregs [198]. In EAMG, we have determined there are numerous benefits from GM-CSF treatment, namely, increasing the number of Tregs, halting antigen-specific T cell proliferation, enhancing IL-10 production, suppressing B cell proliferation, reducing anti-AChR antibody production, and reducing expression of IL-6, IL-17, and IFN-γ [67, 199]. We have showed that GM-CSF can induce tolerogenic semi-mature DCs (CD11c+CD8a−) which cause expansion of Treg cells that suppress EAMG [200]. Such GM-CSF induced Tregs likely suppressed autoimmunity through the secretion of IL-10, as we have shown in an animal model of thyroiditis [201]. Ex vivo co-culture of GM-CSF exposed bone marrow DCs with CD4+ T cells induced selective expansion of Tregs via OX40L and Jagged-1-mediated signaling [200, 202]. Alternatively, it has also been shown that GM-CSF can directly bind to its receptor expressed on human Tregs leading to their expansion [203]. In all, GM-CSF treatment may rectify multiple Treg abnormalities (Table 3).
Inhibit B and TFH cells at GCs
The thymus plays a critical role in anti-AChR antibody production and MG pathogenesis [204]. B cells are implicated not only because of autoantibodies but also because of increases in CXCL13, CCL21, and BAFF in the thymus of MG patients, all of which lead to B cell activation [205]. CXCL13 is utilized by B cells, TFH, and TRH cells to migrate to GCs using CXCR5. Thus, a therapeutic opportunity would be to increase Treg migration to the GC to suppress activated TFH and B cells. This may be achieved with IL-2/mAb complexes (monoclonal antibody greatly increases IL-2 half-life) that have ameliorated EAMG while reducing CD19+ cells, autoantibody levels, and IFN-γ expression [189]. The effects of IL-2/mAb complexes on Tregs and migration to GCs are better understood through an experiment using chronic graft-versus-host disease (cGVHD) in which administration of IL-2/mAb complexes increased splenic Tregs and TFR while ameliorating cGVHD without increasing TFH [94]. Adoptive transfer of wild type Tregs without IL-2/mAb administration caused a reduction in B cell number in the GC dependent upon CXCR5+ Tregs homing to GCs [94]. These findings in GVHD may be relevant to MG as GCs as well as B and T cells are also implicated in GVHD pathogenesis [206]. A mechanism by which IL-2/mAb may act on Tregs over Teff is through the use of IL-2Rα, CD25. The high affinity Tregs have for IL-2 through their CD25 expression will deplete IL-2 from the surrounding milieu restricting IL-2-dependent Teff activation [91]. It should be noted that IL-2/mAb complexes, when used in mice with acute GVHD, resulted in rapid death of mice within 4 days with concomitant increases in Teff, thus the efficacy of treatment may depend on the level of immune activation [94]. Therefore, using IL-2/mAb complexes may be an effective strategy to activate and proliferate Tregs and/or TFR causing suppression and reduction of TFH and B cells in the GC (Fig. 1; Table 3).
OX40L/Jagged-1 Co-treatment
As stated earlier, GM-CSF treatment induced Treg expansion via OX40L and Jagged-1 release from tolerogenic DCs (Fig. 1) [157, 200]. Considering OX40 is found to be upregulated in MG, and our recently published data shows that soluble OX40L and Jagged-1 co-treatment can delay autoimmune diabetes in NOD mice while increasing Tregs, EAMG could be a successful candidate for OX40L and Jagged-1 co-treatment [157, 158, 207]. More interestingly, CD46, a complement receptor expressed on many cell types, is found to co-stimulate with Jagged-1 leading to Treg activation as well as induction of Tr1 cells [161]. Although the role of Tr1 cells is unknown in MG, their suppressive capabilities have been well documented in other autoimmune models [147]. Thus, CD46 activation via Jagged-1 may provide some novel insights into MG treatment modalities [39]. In conclusion, OX40L and Jagged-1 treatment may lead to an increase in Tregs, or even Tr1 cells and rectify OX40 or CD46 dysregulation in MG (Table 3).
TGF-β
TGF-β is a potent activator of Tregs leading to iTreg generation [208]. Generation of iTregs ex vivo in the presence of TGF-β when cultured with B cells from lupus-prone mice (NZB/W) has been found to induce apoptosis of B cells, reduce B cell activation, and reduce autoantibody levels [208]. Additionally, adoptive transfer of iTregs resulted in a greater reduction of autoantibodies compared to nTregs (not exposed to TGF-β ex vivo) [208]. Ex vivo, the addition of TGF-β to mononuclear cells from MG patients induced suppression of cells autoreactive to AChR [209]. The blocking of complement receptors C3a and C5a has been shown to induce expression of TGF-β and IL-10 leading to generation of iTregs which when adoptively transferred potently suppressed the disease [210]. Other studies have also suggested a link between reduced complement expression with increased Treg numbers [211, 212]. Additionally, TGF-β has been shown to reduce iNOS production, again leading to concomitant Treg proliferation [213]. Taken together, it appears TGF-β could restore immunological imbalance in MG patients.
Conclusions
Current MS and MG treatments have modest long-term efficacy and are often associated with severe side effects that fail to treat the underlying disease. Although current treatments do not tend to act on Tregs, they provide a method for regulating autoimmune activation. The Treg augmentation therapies discussed herein focus on rectifying Treg abnormalities by enhancing Treg suppressive activity and/or numbers, increasing Treg migration, causing Treg-dependent B cell destruction or suppression, or enforcing tolerogenic signals from Tregs. Therapies which increase Treg suppressive activity and migration may be particularly useful for MS, whereas MG Treg augmenting therapies should focus on controlling autoreactive B cells. Finally, because these Treg therapeutics appear to act via different mechanisms (Fig. 1), it might be possible to provide synergistic benefits if combined with other tested pre-clinical treatments in MS or MG.
Abbreviations
- AChR:
-
Acetylcholine receptor
- APC:
-
Antigen presenting cell
- B7AP:
-
B7 antisense peptide
- BPI:
-
Bifunctional peptide inhibitor
- CCL:
-
Chemokine ligand
- CCR:
-
Chemokine receptor
- CNS:
-
Central nervous system
- CSF:
-
Central spinal fluid
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DC:
-
Dendritic cell
- DMT:
-
Disease-modifying treatment
- EAE:
-
Experimental autoimmune encephalomyelitis
- EAMG:
-
Experimental autoimmune myasthenia gravis
- FoxP3:
-
Forkhead box P3
- GBS:
-
Guillain-Barre syndrome
- GC:
-
Germinal center
- GCN2:
-
General control nonderepressible 2
- GM-CSF:
-
Granulocyte macrophage-colony stimulating factor
- GVHD:
-
Graft versus host disease
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IL:
-
Interleukin
- iTr35:
-
Induced IL-35 T-regulatory cell
- iTreg:
-
Induced T-regulatory cell
- MG:
-
Myasthenia gravis
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MS:
-
Multiple sclerosis
- NMJ:
-
Neuromuscular junction
- nTreg:
-
Natural T-regulatory cell
- PLP:
-
Proteolipid protein
- RANK:
-
Receptor activator of nuclear factor-kappa B
- SLE:
-
Systemic lupus erythematosus
- Teff:
-
T-effector cell
- TFH:
-
Helper follicular T cells
- TFR:
-
Regulatory follicular T cells
- TGF-β:
-
Transforming growth factor beta
- Th1:
-
T-helper 1 cell
- Th17:
-
T-helper 17 cell
- TNF:
-
Tumor necrosis factor
- Tr1:
-
T-regulatory 1 cell
- Treg:
-
T-regulatory cell
References
Mastorodemos V, Ioannou M, Verginis P. Cell-based modulation of autoimmune responses in multiple sclerosis and experimental autoimmmune encephalomyelitis: therapeutic implications. Neuroimmunomod. 2015;22:181–95.
Ha JC, Richman DP. Myasthenia gravis and related disorders: Pathology and molecular pathogenesis. Biochim Biophys Acta. 2015;1852:651–7.
Filiou MD, Arefin AS, Moscato P, Graeber MB. ‘Neuroinflammation’ differs categorically from inflammation: transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics. 2014;15:201–12.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis. An epidemiologic study. Arch Ophthalmol. 1972;87:135–9.
Banwell B, et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology. 2009;72:232–9.
Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barre syndrome. J Infect Dis. 1997;176 Suppl 2:S92–8.
Phillips 2nd LH. The epidemiology of myasthenia gravis. Ann N Y Acad Sci. 2003;998:407–12.
Kingwell E, et al. Incidence and prevalence of multiple sclerosis in Europe: a systematic review. BMC Neurol. 2013;13:128.
Saguil A, Kane S, Farnell E. Multiple sclerosis: a primary care perspective. Am Fam Physician. 2014;90:644–52.
Dalakas MC. Biologics and other novel approaches as new therapeutic options in myasthenia gravis: a view to the future. Ann N Y Acad Sci. 2012;1274:1–8.
Matney SE, Huff DR. Diagnosis and treatment of myasthenia gravis. Consult Pharm. 2007;22:239–48.
Scott LJ. Glatiramer acetate: a review of its use in patients with relapsing-remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. CNS Drugs. 2013;27:971–88.
Balandina A, Lécart S, Dartevelle P, Saoudi A, Berrih-Aknin S. Functional defect of regulatory CD4(+)CD25(+) T cells in the thymus of patients with autoimmune myasthenia gravis. Blood. 2005;105:735–41.
Xufre C, Costa M, Roura-Mir C, Codina-Busqueta E, Usero L, Pizarro E, Obiols G, Jaraquemada D, Marti M. Low frequency of GITR+ T cells in ex vivo and in vitro expanded Treg cells from type 1 diabetic patients. Int Immunol. 2013;25:563–74.
Chi LJ, Wang HB, Zhang Y, Wang WZ. Abnormality of circulating CD4(+)CD25(+) regulatory T cell in patients with Guillain-Barre syndrome. J Neuroimmunol. 2007;192:206–14.
Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, McCormick TS, Cooper KD. Dysfunctional blood and target tissue CD4 + CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–73.
Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–26.
Basiri K, Etemadifar M, Maghzi AH, Zarghami N. Frequency of myasthenia gravis in multiple sclerosis: Report of five cases from Isfahan. Iran Neurol India. 2009;57:638–40.
Isbister CM, Mackenzie PJ, Anderson D, Wade NK, Oger J. Co-occurrence of multiple sclerosis and myasthenia gravis in British Columbia. Mult Scler. 2003;9:550–3.
Vaknin-Dembinsky A, Abramsky O, Petrou P, Ben-Hur T, Gotkine M, Brill L, Brenner T, Argov Z, Karussis D. Myasthenia gravis-associated neuromyelitis optica-like disease: an immunological link between the central nervous system and muscle? Arch Neurol. 2011;68:1557–61.
Berrih-Aknin S. Myasthenia Gravis: paradox versus paradigm in autoimmunity. J Autoimmun. 2014;52:1–28.
Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74:5–17.
Yi JS, Guidon A, Sparks S, Osborne R, Juel VC, Massey JM, Sanders DB, Weinhold KJ, Guptill JT. Characterization of CD4 and CD8 T cell responses in MuSK myasthenia gravis. J Autoimmun. 2014;52:130–8.
Li S, ** T, Zhang HL, Yu H, Meng F, Concha Quezada H, Zhu J. Circulating Th17, Th22, and Th1 Cells Are Elevated in the Guillain-Barré Syndrome and Downregulated by IVIg Treatments. Mediators Inflamm. 2014;2014:740947.
Link J. Interferon-gamma, interleukin-4 and transforming growth factor-beta mRNA expression in multiple sclerosis and myasthenia gravis. Acta Neurol Scand Suppl. 1994;158:1–58.
Luchtman DW, Ellwardt E, Larochelle C, Zipp F. IL-17 and related cytokines involved in the pathology and immunotherapy of multiple sclerosis: Current and future developments. Cytokine Growth Factor Rev. 2014;25:403–13.
Astier AL, Hafler DA. Abnormal Tr1 differentiation in multiple sclerosis. J Neuroimmunol. 2007;191:70–8.
Thiruppathi M, Rowin J, Ganesh B, Sheng JR, Prabhakar BS, Meriggioli MN. Impaired regulatory function in circulating CD4(+)CD25(high)CD127(low/-) T cells in patients with myasthenia gravis. Clin Immunol. 2012;145:209–23.
Romme Christensen J, Bornsen L, Ratzer R, Piehl F, Khademi M, Olsson T, Sorensen PS, Sellebjerg F. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One. 2013;8:e57820.
Ferguson TB, Clifford DB, Montgomery EB, Bruns KA, McGregor PJ, Trotter JL. Thymectomy in multiple sclerosis. Two preliminary trials. J Thorac Cardiovasc Surg. 1983;85:88–93.
Trotter JL, Clifford DB, Montgomery EB, Ferguson TB. Thymectomy in multiple sclerosis: a 3-year follow-up. Neurology. 1985;35:1049–51.
Haegert DG, Hackenbroch JD, Duszczyszyn D, Fitz-Gerald L, Zastepa E, Mason H, Lapierre Y, Antel J, Bar-Or A. Reduced thymic output and peripheral naive CD4 T-cell alterations in primary progressive multiple sclerosis (PPMS). J Neuroimmunol. 2011;233:233–9.
Vrolix K, et al. Clonal heterogeneity of thymic B cells from early-onset myasthenia gravis patients with antibodies against the acetylcholine receptor. J Autoimmun. 2014;52:101–12.
Fan X, Lin C, Han J, Jiang X, Zhu J, ** T. Follicular Helper CD4+ T Cells in Human Neuroautoimmune Diseases and Their Animal Models. Mediators Inflamm. 2015;2015:638968.
Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161–9.
Utsugisawa K, Nagane Y, Suzuki S, Kondoh R. Antigen-specific T-cell activation in hyperplastic thymus in myasthenia gravis. Muscle Nerve. 2007;36:100–3.
Lee DH, Linker RA. The role of myelin oligodendrocyte glycoprotein in autoimmune demyelination: a target for multiple sclerosis therapy? Expert Opin Ther Targets. 2012;16:451–62.
Fraussen J, de Bock L, Somers V. B cells and antibodies in progressive multiple sclerosis: Contribution to neurodegeneration and progression. Autoimmun Rev. 2016;15:896–9.
Leite MI, et al. Myasthenia gravis thymus: complement vulnerability of epithelial and myoid cells, complement attack on them, and correlations with autoantibody status. Am J Pathol. 2007;171:893–905.
Stuve O, Cepok S, Elias B, Saleh A, Hartung HP, Hemmer B, Kieseier BC. Clinical stabilization and effective B-lymphocyte depletion in the cerebrospinal fluid and peripheral blood of a patient with fulminant relapsing-remitting multiple sclerosis. Arch Neurol. 2005;62:1620–3.
Peres J, Martins R, Alves JD, Valverde A. Rituximab in generalized myasthenia gravis: Clinical, quality of life and cost–utility analysis. Porto Biomed J. 2017;2:81–5.
Batocchi AP, Evoli A, Servidei S, Palmisani MT, Apollo F, Tonali P. Myasthenia gravis during interferon alfa therapy. Neurology. 1995;45:382–3.
Speciale L, Saresella M, Caputo D, Ruzzante S, Mancuso R, Calvo MG, Guerini FR, Ferrante P. Serum auto antibodies presence in multiple sclerosis patients treated with beta-interferon 1a and 1b. J Neurovirol. 2000;6 Suppl 2:S57–61.
Frese A, Bethke F, Ludemann P, Stogbauer F. Development of myasthenia gravis in a patient with multiple sclerosis during treatment with glatiramer acetate. J Neurol. 2000;247:713.
Kaltsonoudis E, Voulgari PV, Konitsiotis S, Drosos AA. Demyelination and other neurological adverse events after anti-TNF therapy. Autoimmun Rev. 2014;13:54–8.
Rowin J, Meriggioli MN, Tuzun E, Leurgans S, Christadoss P. Etanercept treatment in corticosteroid-dependent myasthenia gravis. Neurology. 2004;63:2390–2.
Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–59.
Walker LS. Regulatory T cells overturned: the effectors fight back. Immunology. 2009;126:466–74.
Wehrens EJ, Vastert SJ, Mijnheer G, Meerding J, Klein M, Wulffraat NM, Prakken BJ, van Wijk F. Anti-tumor necrosis factor alpha targets protein kinase B/c-Akt-induced resistance of effector cells to suppression in juvenile idiopathic arthritis. Arthritis Rheum. 2013;65:3279–84.
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4 + CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9.
Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, Buckner JH. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. 2013;5:170ra15.
Schloder J, Berges C, Luessi F, Jonuleit H. Dimethyl Fumarate Therapy Significantly Improves the Responsiveness of T Cells in Multiple Sclerosis Patients for Immunoregulation by Regulatory T Cells. Int J Mol Sci. 2017;18:271.
Gradolatto A, Nazzal D, Truffault F, Bismuth J, Fadel E, Foti M, Berrih-Aknin S. Both Treg cells and Tconv cells are defective in the Myasthenia gravis thymus: roles of IL-17 and TNF-alpha. J Autoimmun. 2014;52:53–63.
Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56.
International Multiple Sclerosis Genetics, C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–60.
Renton AE, et al. A genome-wide association study of myasthenia gravis. JAMA Neurol. 2015;72:396–404.
Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1beta and IL-2 convert human Treg into T(H)17 cells. Clin Immunol. 2009;131:298–307.
Aricha R, Feferman T, Fuchs S, Souroujon MC. Ex Vivo Generated Regulatory T Cells Modulate Experimental Autoimmune Myasthenia Gravis. J Immunol. 2008;180:2132–9.
Aricha R, Reuveni D, Fuchs S, Souroujon MC. Suppression of experimental autoimmune myasthenia gravis by autologous T regulatory cells. J Autoimmun. 2016;67:57–64.
Battaglia A, Di Schino C, Fattorossi A, Scambia G, Evoli A. Circulating CD4 + CD25+ T regulatory and natural killer T cells in patients with myasthenia gravis: a flow cytometry study. J Biol Regul Homeost Agents. 2005;19:54–62.
Noori-Zadeh A, Mesbah-Namin SA, Bistoon-Beigloo S, Bakhtiyari S, Abbaszadeh HA, Darabi S, Rajabibazl M, Abdanipour A. Regulatory T cell number in multiple sclerosis patients: A meta-analysis. Mult Scler Relat Disord. 2016;5:73–6.
Wang XB, Kakoulidou M, Giscombe R, Qiu Q, Huang D, Pirskanen R, Lefvert AK. Abnormal expression of CTLA-4 by T cells from patients with myasthenia gravis: effect of an AT-rich gene sequence. J Neuroimmunol. 2002;130:224–32.
Cohen-Kaminsky S, Levasseur P, Binet JP, Berrih-Aknin S. Evidence of enhanced recombinant interleukin-2 sensitivity in thymic lymphocytes from patients with myasthenia gravis: possible role in autoimmune pathogenesis. J Neuroimmunol. 1989;24:75–85.
Wang WJ, Hao CF, Qu QL, Wang X, Qiu LH, Lin QD. The deregulation of regulatory T cells on interleukin-17-producing T helper cells in patients with unexplained early recurrent miscarriage. Hum Reprod. 2010;25:2591–6.
van Mierlo GJ, Scherer HU, Hameetman M, Morgan ME, Flierman R, Huizinga TW, Toes RE. Cutting edge: TNFR-shedding by CD4 + CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J Immunol. 2008;180:2747–51.
Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4 + CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107:3925–32.
Sheng JR, Muthusamy T, Prabhakar BS, Meriggioli MN. GM-CSF-induced regulatory T cells selectively inhibit anti-acetylcholine receptor-specific immune responses in experimental myasthenia gravis. J Neuroimmunol. 2011;240-241:65–73.
Diaz-Manera J, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–93.
Gertel-Lapter S, Mizrachi K, Berrih-Aknin S, Fuchs S, Souroujon MC. Impairment of regulatory T cells in myasthenia gravis: studies in an experimental model. Autoimmun Rev. 2013;12:894–903.
Li X, **ao BG, ** JY, Lu CZ, Lu JH. Decrease of CD4(+)CD25(high)Foxp3(+) regulatory T cells and elevation of CD19(+)BAFF-R(+) B cells and soluble ICAM-1 in myasthenia gravis. Clin Immunol. 2008;126:180–8.
Dalakas MC. Invited article: inhibition of B cell functions: implications for neurology. Neurology. 2008;70:2252–60.
Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 Controls B cell Responses by Modulating T Follicular Helper, T Follicular Regulatory and T Regulatory Cells. Immunity. 2014;41:1026–39.
Galimberti D, et al. Gender-specific influence of the chromosome 16 chemokine gene cluster on the susceptibility to Multiple Sclerosis. J Neurol Sci. 2008;267:86–90.
Saito R, Onodera H, Tago H, Suzuki Y, Shimizu M, Matsumura Y, Kondo T, Itoyama Y. Altered expression of chemokine receptor CXCR5 on T cells of myasthenia gravis patients. J Neuroimmunol. 2005;170:172–8.
Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–73.
Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, Barthlott T, Stockinger B, Wraith DC, O’Garra A. IL-10-Secreting Regulatory T Cells Do Not Express Foxp3 but Have Comparable Regulatory Function to Naturally Occurring CD4 + CD25+ Regulatory T Cells. J Immunol. 2004;172:5986–93.
Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol. 2009;21:1105–11.
Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10.
Lühder F, Höglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T Lymphocyte–associated Antigen 4 (CTLA-4) Regulates the Unfolding of Autoimmune Diabetes. J Exp Med. 1998;187:427–32.
Collison LW, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11:1093–101.
Masli S, Turpie B. Anti-inflammatory effects of tumour necrosis factor (TNF)-alpha are mediated via TNF-R2 (p75) in tolerogenic transforming growth factor-beta-treated antigen-presenting cells. Immunology. 2009;127:62–72.
Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–30.
Suzuki H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–6.
Tai X, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood. 2012;119:5155–63.
Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-β: the role of T regulatory cells. Immunology. 2006;117:433–42.
Olson B, Sullivan J, Burlingham W. Interleukin 35: A Key Mediator of Suppression and the Propagation of Infectious Tolerance. Front Immunol. 2013;4:315.
Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44:316–29.
Hall BM, Tran GT, Robinson CM, Hodgkinson SJ. Induction of antigen specific CD4(+)CD25(+)Foxp3(+)T regulatory cells from naive natural thymic derived T regulatory cells. Int Immunopharmacol. 2015;28:875–86.
Ma J, et al. Differential role of all-trans retinoic acid in promoting the development of CD4+ and CD8+ regulatory T cells. J Leukoc Biol. 2014;95:275–83.
McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77:1–10.
Letourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. 2009;123:758–62.
Iikuni N, Lourenco EV, Hahn BH, La Cava A. Cutting edge: Regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol. 2009;183:1518–22.
Wen Y, Yang B, Lu J, Zhang J, Yang H, Li J. Imbalance of circulating CD4(+)CXCR5(+)FOXP3(+) Tfr-like cells and CD4(+)CXCR5(+)FOXP3(-) Tfh-like cells in myasthenia gravis. Neurosci Lett. 2016;630:176–82.
McDonald-Hyman C, et al. Therapeutic regulatory T-cell adoptive transfer ameliorates established murine chronic GVHD in a CXCR5-dependent manner. Blood. 2016;128:1013–7.
Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, Hohlfeld R, Wekerle H. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. 2014;54:33–50.
Haider L, Zrzavy T, Hametner S, Hoftberger R, Bagnato F, Grabner G, Trattnig S, Pfeifenbring S, Bruck W, Lassmann H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. 2016;139:807–15.
Chung DT, Korn T, Richard J, Ruzek M, Kohm AP, Miller S, Nahill S, Oukka M. Anti-thymocyte globulin (ATG) prevents autoimmune encephalomyelitis by expanding myelin antigen-specific Foxp3+ regulatory T cells. Int Immunol. 2007;19:1003–10.
Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012;123:627–38.
Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of Multiple Sclerosis. Eur J Pharmacol. 2015;759:182–91.
Mirshafiey A, M Kianiaslani. Autoantigens and autoantibodies in multiple sclerosis. Iran J Allergy Asthma Immunol. 2013;12:292–303.
van Noort JM, Bsibsi M, Nacken PJ, Verbeek R, Venneker EH. Therapeutic Intervention in Multiple Sclerosis with Alpha B-Crystallin: A Randomized Controlled Phase IIa Trial. PLoS One. 2015;10:e0143366.
Kerlero de Rosbo N, et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol. 1997;27:3059–69.
Ayoglu B, Haggmark A, Khademi M, Olsson T, Uhlen M, Schwenk JM, Nilsson P. Autoantibody profiling in multiple sclerosis using arrays of human protein fragments. Mol Cell Proteomics. 2013;12:2657–72.
Fritzsching B, Haas J, Konig F, Kunz P, Fritzsching E, Poschl J, Krammer PH, Bruck W, Suri-Payer E, Wildemann B. Intracerebral human regulatory T cells: analysis of CD4+ CD25+ FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS One. 2011;6:e17988.
de Oliveira DM, de Oliveira EM, Ferrari Mde F, Semedo P, Hiyane MI, Cenedeze MA, Pacheco-Silva A, Camara NO, Peron JP. Simvastatin ameliorates experimental autoimmune encephalomyelitis by inhibiting Th1/Th17 response and cellular infiltration. Inflammopharmacol. 2015;23:343–54.
Muller M, et al. CXCR3 Signaling Reduces the Severity of Experimental Autoimmune Encephalomyelitis by Controlling the Parenchymal Distribution of Effector and Regulatory T Cells in the Central Nervous System. J Immunol. 2007;179:2774–86.
Almolda B, Gonzalez B, Castellano B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J Neuroimmunol. 2010;223:39–54.
Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302.
Weber MS, Prod’homme T, Youssef S, Dunn SE, Steinman L, Zamvil SS. Neither T-helper type 2 nor Foxp3+ regulatory T cells are necessary for therapeutic benefit of atorvastatin in treatment of central nervous system autoimmunity. J Neuroinflammation. 2014;11:29.
Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, Cullimore ML, Rostami A, Xu H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol. 2010;185:5953–61.
Kwilasz AJ, Grace PM, Serbedzija P, Maier SF, Watkins LR. The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacol. 2015;96:55–69.
Beebe AM, Cua DJ, de Waal Malefyt R. The role of interleukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev. 2002;13:403–12.
Liu Y, et al. FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med. 2014;20:272–82.
Tauro S, Nguyen P, Li B, Geiger TL. Diversification and senescence of Foxp3+ regulatory T cells during experimental autoimmune encephalomyelitis. Eur J Immunol. 2013;43:1195–207.
McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4 + CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–32.
Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. 1998;161:3299–306.
Korn T, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–31.
Keil M, Sonner JK, Lanz TV, Oezen I, Bunse T, Bittner S, Meyer HV, Meuth SG, Wick W, Platten M. General control non-derepressible 2 (GCN2) in T cells controls disease progression of autoimmune neuroinflammation. J Neuroimmunol. 2016;297:117–26.
Butti E, et al. IL4 gene delivery to the CNS recruits regulatory T cells and induces clinical recovery in mouse models of multiple sclerosis. Gene Ther. 2008;15:504–15.
Yang G, Li H, Yao Y, Xu F, Bao Z, Zhou J. Treg/Th17 imbalance in malignant pleural effusion partially predicts poor prognosis. Oncol Rep. 2015;33:478–84.
Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286–93.
Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, Barsheshet Y, Karp CL, Karin N. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest. 2014;124:2009–22.
Sorensen TL, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. 2002;127:59–68.
Hall AO, et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–23.
Ben-Zacharia AB. Therapeutics for Multiple Sclerosis Symptoms. Mt Sinai J Med. 2011;78:176–91.
Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2–13.
La Mantia L, Munari LM, Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst Rev. 2010;5:Cd004678.
Clanet MC, Wolinsky JS, Ashton RJ, Hartung HP, Reingold SC. Risk evaluation and monitoring in multiple sclerosis therapeutics. Mult Scler. 2014;20:1306–11.
Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275:350–63.
Pfender N, Martin R. Daclizumab (anti-CD25) in multiple sclerosis. Exp Neurol. 2014;262(Pt A):44–51.
Chen M, Chen G, Deng S, Liu X, Hutton GJ, Hong J. IFN-beta induces the proliferation of CD4 + CD25 + Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J Neuroimmunol. 2012;242:39–46.
Haas J, Schwarz A, Korporal-Kunke M, Jarius S, Wiendl H, Kieseier BC, Wildemann B. Fingolimod does not impair T-cell release from the thymus and beneficially affects Treg function in patients with multiple sclerosis. Mult Scler. 2015;21:1521–32.
Braitch M, Harikrishnan S, Robins RA, Nichols C, Fahey AJ, Showe L, Constantinescu CS. Glucocorticoids increase CD4CD25 cell percentage and Foxp3 expression in patients with multiple sclerosis. Acta Neurol Scand. 2009;119:239–45.
Olsen PC, Kitoko JZ, Ferreira TP, de-Azevedo CT, Arantes AC, Martins Mu A. Glucocorticoids decrease Treg cell numbers in lungs of allergic mice. Eur J Pharmacol. 2015;747:52–8.
Putzki N, Kumar M, Kreuzfelder E, Grosse-Wilde H, Diener HC, Limmroth V. Mitoxantrone does not restore the impaired suppressive function of natural regulatory T cells in patients suffering from multiple sclerosis. A longitudinal ex vivo and in vitro study. Eur Neurol. 2009;61:27–32.
Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of natalizumab on circulating B cells, T regulatory cells and natural killer cells. Eur Neurol. 2010;63:311–7.
Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult Scler. 2016;22:1061–70.
Ochoa-Repáraz J, Colpitts SL, Kircher C, Kasper EJ, Telesford KM, Begum-Haque S, Pant A, Kasper LH. Induction of gut regulatory CD39(+) T cells by teriflunomide protects against EAE. Neurol Neuroimmunol Neuroinflamm. 2016;3:e291.
Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J Neuroimmunol. 2009;216:113–7.
Wust S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, Luhder F, Reichardt HM. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol. 2008;180:8434–43.
Srinivasan M, Gienapp IE, Stuckman SS, Rogers CJ, Jewell SD, Kaumaya PTP, Whitacre CC. Suppression of Experimental Autoimmune Encephalomyelitis Using Peptide Mimics of CD28. J Immunol. 2002;169:2180–8.
Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, de Waal-Malefyt R, Coffman RL, Hawrylowicz CM, O’Garra A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–16.
Sloane E, et al. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental Multiple Sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain Behav Immun. 2009;23:92–100.
Colombel JF, et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut. 2001;49:42–6.
Saxena A, Khosraviani S, Noel S, Mohan D, Donner T, Hamad AR. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine. 2015;74:27–34.
Mari ER, Rasouli J, Ciric B, Moore JN, Conejo-Garcia JR, Rajasagi N, Zhang GX, Rabinovich GA, Rostami A. Galectin-1 is essential for the induction of MOG35-55 -based intravenous tolerance in experimental autoimmune encephalomyelitis. Eur J Immunol. 2016;46:1783–96.
Wu C, et al. Metallothioneins negatively regulate IL-27–induced type 1 regulatory T-cell differentiation. Proc Natl Acad Sci. 2013;110:7802–7.
Banchereau J, Pascual V, O’Garra A. From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nat Immunol. 2012;13:925–31.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9.
Choi J, Leung PS, Bowlus C, Gershwin ME. IL-35 and Autoimmunity: a Comprehensive Perspective. Clin Rev Allergy Immunol. 2015;49:327–32.
Bardel E, Larousserie F, Charlot-Rabiega P, Coulomb-L’Hermine A, Devergne O. Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J Immunol. 2008;181:6898–905.
Iwasaki Y, Fujio K, Okamura T, Yamamoto K. Interleukin-27 in T Cell Immunity. Int J Mol Sci. 2015;16:2851–63.
Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang GX, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–75.
Apetoh L, Quintana FJ, Pot C, Joller N, **ao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK. The Aryl hydrocarbon Receptor (AhR) interacts with c-Maf to promote the differentiation of IL-27-induced regulatory type 1 (T(R)1) cells. Nat Immunol. 2010;11:854–61.
Badawi AH, P Kiptoo, TJ. Siahaan. Immune Tolerance Induction against Experimental Autoimmune Encephalomyelitis (EAE) Using A New PLP-B7AP Conjugate that Simultaneously Targets B7/CD28 Costimulatory Signal and TCR/MHC-II Signal. J Mult Scler. 2015;2:1–10.
Manikwar P, Kiptoo P, Badawi AH, Buyuktimkin B, Siahaan TJ. Antigen-specific blocking of CD4-specific immunological synapse formation using BPI and current therapies for autoimmune diseases. Med Res Rev. 2012;32:727–64.
Gopisetty A, Bhattacharya P, Haddad C, Bruno Jr JC, Vasu C, Miele L, Prabhakar BS. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol. 2013;190:5516–25.
Kumar P, Alharshawi K, Bhattacharya P, Marinelarena A, Haddad C, Sun Z, Chiba S, Epstein AL, Prabhakar BS. Soluble OX40L and JAG1 Induce Selective Proliferation of Functional Regulatory T-Cells Independent of canonical TCR signaling. Sci Rep. 2017;7:39751.
Elyaman W, Bradshaw EM, Wang Y, Oukka M, Kivisakk P, Chiba S, Yagita H, Khoury SJ. JAGGED1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5990–8.
Stidworthy MF, Genoud S, Li WW, Leone DP, Mantei N, Suter U, Franklin RJ. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain. 2004;127:1928–41.
Le Friec G. The CD46 and Jagged1 interaction is critical for human T helper 1 immunity. Nat Immunol. 2012;13:1213–21.
Ma A, et al. Dysfunction of IL-10-producing type 1 regulatory T cells and CD4(+)CD25(+) regulatory T cells in a mimic model of human multiple sclerosis in Cynomolgus monkeys. Int Immunopharmacol. 2009;9:599–608.
Ni Choileain S, Astier AL. CD46 plasticity and its inflammatory bias in multiple sclerosis. Arch Immunol Ther Exp. 2011;59:49–59.
Torok K, Dezso B, Bencsik A, Uzonyi B, Erdei A. Complement receptor type 1 (CR1/CD35) expressed on activated human CD4+ T cells contributes to generation of regulatory T cells. Immunol Lett. 2015;164:117–24.
Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–92.
Haddad CS, Bhattacharya P, Alharshawi K, Marinelarena A, Kumar P, El-Sayed O, Elshabrawy HA, Epstein AL, Prabhakar BS. Age-dependent divergent effects of OX40L treatment on the development of diabetes in NOD mice. Autoimmunity. 2016;49:298–311.
Ruby CE, Yates MA, Hirschhorn-Cymerman D, Chlebeck P, Wolchok JD, Houghton AN, Offner H, Weinberg AD. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol. 2009;183:4853–7.
Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, Guillemin GJ. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. 2012;72:5649–57.
Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014;63:721–35.
Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;129:186–96.
Haas J, et al. Specific recruitment of regulatory T cells into the CSF in lymphomatous and carcinomatous meningitis. Blood. 2008;111:761–6.
Ehirchiou D, Muller YD, Chicheportiche R, Heyrani Nobari R, Madelon N, Schneider MK, Seebach JD. Chemoattractant Signals and Adhesion Molecules Promoting Human Regulatory T Cell Recruitment to Porcine Endothelium. Transplantation. 2016;100:753–62.
Tüzün E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev. 2013;12:904–11.
Evoli A, Padua L. Diagnosis and therapy of myasthenia gravis with antibodies to muscle-specific kinase. Autoimmun Rev. 2013;12:931–5.
Zisimopoulou P, Brenner T, Trakas N, Tzartos SJ. Serological diagnostics in myasthenia gravis based on novel assays and recently identified antigens. Autoimmun Rev. 2013;12:924–30.
Diaz A, Black E, Dunning J. Is thymectomy in non-thymomatous myasthenia gravis of any benefit? Interact Cardiovasc Thorac Surg. 2014;18:381–9.
Losen M, Martinez-Martinez P, Molenaar PC, Lazaridis K, Tzartos S, Brenner T, Duan RS, Luo J, Lindstrom J, Kusner L. Standardization of the experimental autoimmune myasthenia gravis (EAMG) model by immunization of rats with Torpedo californica acetylcholine receptors--Recommendations for methods and experimental designs. Exp Neurol. 2015;270:18–28.
Meinl E, Klinkert WE, Wekerle H. The thymus in myasthenia gravis. Changes typical for the human disease are absent in experimental autoimmune myasthenia gravis of the Lewis rat. Am J Pathol. 1991;139:995–1008.
Lindstrom JM. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453–77.
Fuchs S, Aricha R, Reuveni D, Souroujon MC. Experimental Autoimmune Myasthenia Gravis (EAMG): from immunochemical characterization to therapeutic approaches. J Autoimmun. 2014;54:51–9.
Huang YM, Pirskanen R, Giscombe R, Link H, Lefvert AK. Circulating CD4 + CD25+ and CD4 + CD25+ T cells in myasthenia gravis and in relation to thymectomy. Scand J Immunol. 2004;59:408–14.
Masuda M, Matsumoto M, Tanaka S, Nakajima K, Yamada N, Ido N, Ohtsuka T, Nishida M, Hirano T, Utsumi H. Clinical implication of peripheral CD4 + CD25+ regulatory T cells and Th17 cells in myasthenia gravis patients. J Neuroimmunol. 2010;225:123–31.
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–53.
Totsuka T, et al. RANK-RANKL signaling pathway is critically involved in the function of CD4 + CD25+ regulatory T cells in chronic colitis. J Immunol. 2009;182:6079–87.
Wang J, Yu L, Jiang C, Fu X, Liu X, Wang M, Ou C, Cui X, Zhou C, Wang J. Cerebral ischemia increases bone marrow CD4 + CD25 + FoxP3+ regulatory T cells in mice via signals from sympathetic nervous system. Brain Behav Immun. 2015;43:172–83.
Wei S, Kryczek I, Zou W. Regulatory T-cell compartmentalization and trafficking. Blood. 2006;108:426–31.
Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol. 2014;92:64–71.
Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–82.
Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi FD. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur J Immunol. 2010;40:1577–89.
Rocha JA, Ribeiro SP, Franca CM, Coelho O, Alves G, Lacchini S, Kallas EG, Irigoyen MC, Consolim-Colombo FM. Increase in cholinergic modulation with pyridostigmine induces anti-inflammatory cell recruitment soon after acute myocardial infarction in rats. Am J Physiol Regul Integr Comp Physiol. 2016;310:R697–706.
Li Z, Mou W, Lu G, Cao J, He X, Pan X, Xu K. Low-dose rituximab combined with short-term glucocorticoids up-regulates Treg cell levels in patients with immune thrombocytopenia. Int J Hematol. 2011;93:91–8.
Maddur MS, Othy S, Hegde P, Vani J, Lacroix-Desmazes S, Bayry J, Kaveri SV. Immunomodulation by intravenous immunoglobulin: role of regulatory T cells. J Clin Immunol. 2010;30 Suppl 1:S4–8.
Tselios K, Sarantopoulos A, Gkougkourelas I, Boura P. The influence of therapy on CD4 + CD25(high)FOXP3+ regulatory T cells in systemic lupus erythematosus patients: a prospective study. Scand J Rheumatol. 2015;44:29–35.
Cooper V, Metcalf L, Versnel J, Upton J, Walker S, Horne R. Patient-reported side effects, concerns and adherence to corticosteroid treatment for asthma, and comparison with physician estimates of side-effect prevalence: a UK-wide, cross-sectional study. NPJ Prim Care Respir Med. 2015;25:15026.
Aung T, Dowden AY. Successful desensitization protocol for pyridostigmine hypersensitivity in a patient with myasthenia gravis. Ann Allergy Asthma Immunol. 2013;110:308.
Gupta A, Goyal V, Srivastava AK, Shukla G, Behari M. Remission And relapse of myasthenia gravis on long-term azathioprine: An ambispective study. Muscle Nerve. 2016;54:405–12.
Wolfe GI, et al. Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med. 2016;375:511–22.
Rowin J, Thiruppathi M, Arhebamen E, Sheng J, Prabhakar BS, Meriggioli MN. Granulocyte macrophage colony-stimulating factor treatment of a patient in myasthenic crisis: effects on regulatory T cells. Muscle Nerve. 2012;46:449–53.
Sheng JR, Li LC, Ganesh BB, Prabhakar BS, Meriggioli MN. Regulatory T cells induced by GM-CSF suppress ongoing experimental myasthenia gravis. Clin Immunol. 2008;128:172–80.
Sheng JR, Li L, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of Experimental Autoimmune Myasthenia Gravis by Granulocyte-Macrophage Colony-Stimulating Factor Is Associated with an Expansion of FoxP3+ Regulatory T Cells. J Immunol. 2006;177:5296–306.
Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c + CD8a--dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol. 2009;21:269–82.
Bhattacharya P, Gopisetty A, Ganesh BB, Sheng JR, Prabhakar BS. GM-CSF-induced, bone-marrow-derived dendritic cells can expand natural Tregs and induce adaptive Tregs by different mechanisms. J Leukoc Biol. 2011;89:235–49.
Kared H, Leforban B, Montandon R, Renand A, Layseca Espinosa E, Chatenoud L, Rosenstein Y, Schneider E, Dy M, Zavala F. Role of GM-CSF in tolerance induction by mobilized hematopoietic progenitors. Blood. 2008;112:2575–8.
Cavalcante P, Le Panse R, Berrih-Aknin S, Maggi L, Antozzi C, Baggi F, Bernasconi P, Mantegazza R. The thymus in myasthenia gravis: Site of “innate autoimmunity”? Muscle Nerve. 2011;44:467–84.
Berrih-Aknin S, Ragheb S, Le Panse R, Lisak RP. Ectopic germinal centers, BAFF and anti-B-cell therapy in myasthenia gravis. Autoimmun Rev. 2013;12:885–93.
Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, von Bergwelt-Baildon MS. The role of B cells in the pathogenesis of graft-versus-host disease. Blood. 2009;114:4919–27.
**aoyan Z, Pirskanen R, Malmstrom V, Lefvert AK. Expression of OX40 (CD134) on CD4+ T-cells from patients with myasthenia gravis. Clin Exp Immunol. 2006;143:110–6.
Xu A, et al. TGF-beta-Induced Regulatory T Cells Directly Suppress B Cell Responses through a Noncytotoxic Mechanism. J Immunol. 2016;196:3631–41.
Link J, He B, Navikas V, Palasik W, Fredrikson S, Soderstrom M, Link H. Transforming growth factor-beta 1 suppresses autoantigen-induced expression of pro-inflammatory cytokines but not of interleukin-10 in multiple sclerosis and myasthenia gravis. J Neuroimmunol. 1995;58:21–35.
Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14:162–71.
Yuan Y, Yan D, Han G, Gu G, Ren J. Complement C3 depletion links to the expansion of regulatory T cells and compromises T-cell immunity in human abdominal sepsis: a prospective pilot study. J Crit Care. 2013;28:1032–8.
Gao X, Liu H, Ding G, Wang Z, Fu H, Ni Z, Ma J, Liu F, Fu Z. Complement C3 deficiency prevent against the onset of streptozotocin-induced autoimmune diabetes involving expansion of regulatory T cells. Clin Immunol. 2011;140:236–43.
Jayaraman P, Alfarano MG, Svider PF, Parikh F, Lu G, Kidwai S, **ong H, Sikora AG. iNOS expression in CD4+ T cells limits Treg induction by repressing TGFbeta1: combined iNOS inhibition and Treg depletion unmask endogenous antitumor immunity. Clin Cancer Res. 2014;20:6439–51.
Acknowledgements
Palash Bhattacharya is acknowledged for critical reading of the manuscript.
Funding
NIH 1RO1AI107516-01A1 and NIH 1R41AI125039-01 from the National Institutes of Health.
Availability of data and materials
Data supporting the conclusions of this article are included within the “References” section.
Authors’ contributions
KD wrote the manuscript, edited by SJ, and BP conceptualized the article and edited the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Danikowski, K.M., Jayaraman, S. & Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation 14, 117 (2017). https://doi.org/10.1186/s12974-017-0892-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-017-0892-8