
Abstract
Bone regeneration therapy is clinically important, and targeted regulation of endoplasmic reticulum (ER) stress is important in regenerative medicine. The processing of proteins in the ER controls cell fate. The accumulation of misfolded and unfolded proteins occurs in pathological states, triggering ER stress. ER stress restores homeostasis through three main mechanisms, including protein kinase-R-like ER kinase (PERK), inositol-requiring enzyme 1ɑ (IRE1ɑ) and activating transcription factor 6 (ATF6), collectively known as the unfolded protein response (UPR). However, the UPR has both adaptive and apoptotic effects. Modulation of ER stress has therapeutic potential for numerous diseases. Repair of bone defects involves both angiogenesis and bone regeneration. Here, we review the effects of ER stress on osteogenesis and angiogenesis, with emphasis on ER stress under high glucose (HG) and inflammatory conditions, and the use of ER stress inducers or inhibitors to regulate osteogenesis and angiogenesis. In addition, we highlight the ability for exosomes to regulate ER stress. Recent advances in the regulation of ER stress mediated osteogenesis and angiogenesis suggest novel therapeutic options for bone defects.
Similar content being viewed by others
Introduction
As the elderly population has increased globally, so has the number of patients with clinical bone defects [1, 2]. Patients with diabetes mellitus have a high incidence of bone defects [30] and accumulation of misfolded or unfolded proteins in the ER, leading to ER stress [31, 32]. Therefore, ER stress is an important cellular defense mechanism and is vital for maintaining ER homeostasis.
ER stress signaling pathways
ER stress can be classified as the UPR, ER overload response, and sterol regulatory cascade [33]. UPR occurs when a signal of misfolded ER proteins is transmitted to the nucleus [34]. Ischemia [30]. However, in ER stress, BiP dissociates and binds unfolded or misfolded proteins and perform protein folding [48], activating ER receptors [49]. The IRE1ɑ-X-box binding protein (XBP1), PERK-eukaryotic initiation factor 2ɑ (eIF2ɑ), and ATF6 signaling pathways induce the UPR and restore ER stability [50, 51]. IRE1ɑ is the most evolutionarily conserved factor in the UPR [30, 38]. Activated PERK phosphorylates eIF2ɑ, attenuating protein translation to relieve the ER load under stress, and promotes ATF4 translation [30, 45]. The PERK signaling pathway is associated with a series of immune metabolic diseases [52, 53], including tumors [30], and full-length ATF6 (ATF6p90) monomer increases and is transferred to the Golgi apparatus, where it is cleaved by the site 1 protease (S1P) and site 2 protease (S2P) to release an N-terminal transcriptionally active 50 kDa fragment (ATF6p50) [38, 57]. ATF6p50 is transported to the nucleus to perform functions such as protein folding [38]. ATF6 also maintains the stability of viral proteins [57] and homeostasis in normally develo** tissues and organs [58].

Copyright© The Authors 2020. Published by Springer Ltd
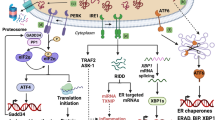
Major UPR pathways initiated in the ER [38]. RIDD: regulated IRE1ɑ-dependent decay; TRAF: tumor necrosis factor receptor associated factor; ERAD: ER-associated protein degradation; PP1: protein phosphatase 1; CreP: constitutive repressor of eIF2ɑ phosphorylation; DR5: death receptor 5; TXNIP: thioredoxin-interacting protein; IP3R: inositol-1,4,5-triphosphate receptor; BI-1: Bax inhibitor-1; GADD34: growth arrest and DNA damage inducible gene 34. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation, and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–38.
ER stress pathways: a double-edged sword
After the occurrence of ER stress, misfolded or unfolded proteins that accumulate in the ER are eliminated through two primary degradation pathways: ER-associated degradation (ERAD) and autophagy [59]. ERAD is activated in response to ER stress, as it maintains ER homeostasis by eliminating misfolded proteins from the ER and preventing their accumulation [60]. The UPR controls cell fate [17, 19, 20, 61]. A prolonged UPR indicates non-recovery from ER stress, and adaptive output cannot compensate for the pressure in the ER, and the UPR induces apoptosis [20]. Sustained activation of ATF4 in combination with CCAAT-enhancer-binding protein homologous protein (CHOP) induces apoptosis [34]. Thus, the dual role of the PERK-eIF2ɑ axis is vital for coordinating translation and protein balance. There are three main mechanisms (Fig. 1): the IRE1ɑ/ASK1 (apoptosis signal regulating kinase 1)/JNK (c-jun kinase) pathway [62], caspase-12-dependent pathway [63], and growth arrest and DNA damage-inducible 153 (CHOP/GADD153) pathway [64, 65]. CHOP, a key apoptotic factor, upregulates ROS, triggers calcium (Ca2+) release, and promotes transcription, constituting a positive-feedback loop that triggers apoptosis [34]. It also downregulates the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) to induce apoptosis [66]. Despite advances in research on the mechanism of ER stress, the balance between pro-survival and pro-death UPR signals remains unclear, and the full extent of ER stress's role in different stages of disease is yet to be fully elucidated. Future research is necessary to answer these key questions.
ER stress pathways in orthopedics
Osteoblasts play a critical role in bone formation and reconstruction by synthesizing new collagen. Because osteoblasts secrete a significant amount of extracellular matrix proteins, they are particularly vulnerable to ER stress-induced dysfunction. Targeted ER stress therapy can be used to treat orthopedic diseases. Liu et al. [67] discovered that IL-1β can induce excessive ER stress in chondrocytes, leading to chondrocyte apoptosis and subsequent cartilage degradation, which accelerates the progression of osteoarthritis (OA). Inhibition of ER stress by the IRE1ɑ pathway suppresses chondrocyte apoptosis, thus mitigating the progression of OA [68]. Sim et al. [69] found that the function of ERAD, which is regulated by ER stress, was reduced in patients with OA, leading to the accumulation of misfolded proteins and cartilage loss. Enhanced ERAD activity is necessary for cartilage formation and maintenance. The activation of PERK and ATF4 is involved in the inducing the stress response protein sestrin2 under ER stress after spinal cord injury (SCI) [70]. Inhibiting ER stress through overexpression of sestrin2 promotes functional recovery and neuronal survival, indicating its potential as a therapeutic target for SCI repair. Huang et al. [71] found that inhibition of neuronal apoptosis mediated by ER stress can reduce apoptosis and protects neurons. ER stress has potential to be a new target for treating SCI; Metastasis of osteosarcoma cells can be inhibited by knocking out secretion-associated Ras-related GTPase 1A (SAR1A), a key regulator of ER homeostasis [72]. Moreover, ER stress can induce hypertrophic chondrocyte dysfunction, which may be a potential cause of osteogenesis imperfecta (OI) [73]. Nevertheless, studies have demonstrated that downstream ER stress is necessary to maintain Ol bone integrity to a certain extent. Reducing ER stress alone may not be sufficient to rescue Ol phenotype and may even exacerbate it [74]. Although we have known that ER stress is part of the mechanism of OI disease, how to improve OI bone strength by regulating ER stress remains to be studied.
The repair and regeneration of bone defects caused by trauma, tumor, infection and other factors have been significant clinical challenges. If ER stress can be precisely regulated to an appropriate level through bone tissue engineering or stem cell therapy, it could help in the rapid regeneration of bone tissue. **ang et al. [75] modulated the expression of osteogenic proteins through the PERK-eIF2ɑ-ATF4 pathway of appropriate ER stress by Ca2+ changes mediated by biphasic calcium phosphate, a classic bone void filler. Zheng et al. [76] used the osteogenesis-promoting drug HA15 to target HSPA5 to inhibit excessive ER stress and ultimately promote osteogenesis and angiogenesis in rabbit bone defect models. Future studies can use the involvement of the ER stress pathway in the regulation of osteogenesis and angiogenesis as a starting point through cell experiments, investigate the precise mechanism by which ER stress regulates osteogenesis and angiogenesis, and seek more possible therapeutic targets and interventions in the pathogenesis of bone defect from the level of gene regulation, bringing good news to the majority of patients.
Effects of ER stress pathways on osteogenesis
Appropriate ER stress contributes to osteogenic differentiation
Bone morphogenetic proteins (BMPs) are implicated in osteogenic differentiation and ectopic bone formation [77]. BMP2 and BMP9 induce ER stress to promote the differentiation of BMSCs into osteoblasts [78,79,80,81]. UPR signaling is an essential regulator of bone development [82, 83].
The three UPR signaling pathways are linked to the promotion of osteogenic differentiation by ER stress. Kazuhisa et al. [84] discovered Osterix (Osx), a transcription factor necessary for bone formation. Ten years later, Stavroula [85] identified Osx as a target gene of XBP1, linking ER stress and osteogenesis. The IRE1ɑ-XBP1 signaling pathway promotes not only osteoblast maturation by promoting Osx transcription [86] but also bone regeneration via myostatin mRNA decay [87]. ATF4 is a key transcription factor for osteoblast differentiation and bone formation [88, 89]. Activation of the PERK-eIF2ɑ-ATF4 pathway promotes the expression of genes required for osteogenesis [90] and induces osteogenic differentiation [91, 92] and type I collagen secretion, which are essential for neonatal bone development and osteogenic differentiation [93]. Won-Gu et al. [94] showed that BMP2 stimulates osteoblast differentiation by regulating osteocalcin gene expression via the ER stress-activated ATF6 pathway [58]. Although the three UPR signaling pathways are implicated in osteogenesis, the underlying mechanisms are unclear.
Excessive ER stress induces osteoblast apoptosis
Excessive ER stress inhibits osteogenic differentiation and induces their apoptosis [91, 95, 96], which is an important mechanism of osteoporosis [97]. The effect may be related to the overexpression of CHOP caused by excessive ER stress [98], and there are sex differences in sensitivity to CHOP [99]. Overexpression of CHOP reduces alkaline phosphatase activity and calcified bone nodule formation [100], and initiates osteoblast apoptosis, inhibits bone formation, and induces osteopenia [98, 100].
ER stress-mediated osteoblast apoptosis is driven by an increase in the intracellular Ca2+ concentration [101]. An increased intracellular Ca2+ disrupts Ca2+ homeostasis, leading to Ca2+ overload [102] and excessive ER stress103 and inducing osteoblast apoptosis [104, 105]. Furthermore, micronutrients such as cadmium [106], fluorine [107, 108], and iron [109] initiate the ER stress apoptosis pathway by increasing intracellular Ca2+. Therefore, controlling intracellular Ca2+ has therapeutic potential for micronutrient-induced osteoporosis. We summarize the effects of ER stress inducers on osteogenic differentiation in Table 1.
Regulation of ER stress pathways to interfere with osteogenesis
GCs induce osteoblast apoptosis by activating ER stress pathways
A normal concentration of glucose does not activate ER stress [110], but chronic HG induces pancreatic β cells to continuously secrete Ca2+ to activate ER stress [37], thus inhibiting osteogenic differentiation in a glucose concentration-dependent manner [110].
Since 1984, glucocorticoids (GCs) have been used for variety of immune-related diseases [111]. However, long-term use of GCs increases the incidence of osteonecrosis, among which osteonecrosis of the femoral head (ONFH) is the most common [112]. Although the mechanism of GC-induced ONFH is unclear, GCs can activate ER stress and promote the production of ROS, thereby inducing apoptosis in osteoblasts [113, 114], particularly in the proximal femur [115]. This may be a mechanism of ONFH.
The PERK-eIF2ɑ-ATF4-CHOP pathway is implicated in GC-induced osteoblast apoptosis [116]. Therefore, controlling this pathway could ameliorate GC-induced osteoblast apoptosis. The plant compound geniposide (GEN) [117], 4-phenylbutyric acid (4-PBA) [117, 118], the PERK phosphorylation inhibitor GSK2656157 [116], and melatonin [119] can block PERK downstream signaling and significantly inhibit ER stress, thereby attenuating GC-induced osteoblast apoptosis. GEN together with the plant compound paeoniflorin activate autophagy in vivo and in vitro, thus suppressing GC-induced apoptosis [115, 120]. 4-PBA downregulates ATF4 and reduces mutant type I collagen [121], whereas salubrinal (inhibitor of eIF2ɑ dephosphorylation) upregulates ATF4 [35, 122]. Both regulate the eIF2ɑ pathway, thereby reducing ER stress to promote osteogenesis. Unfortunately, salubrinal has no effect on osteoblast apoptosis induced by high-dose GC [114].
Regulation of ER stress pathways on osteogenesis under inflammatory conditions
Long-term inflammatory responses can affect stem cells' ability to repair [123]. Tumor necrosis factor ɑ (TNF-ɑ)-induced inflammation has been reported to inhibit osteogenic differentiation of BMSCs [124], possibly because ER stress-activated nuclear factor κB (NF-κB) translocates into the nucleus to promote the transcription of other pro-inflammatory cytokines [125] and osteolysis [126]. Xue et al. [96] found for the first time that long-term chronic inflammation reduces the expression of lysine acetyltransferase 6B (KAT6B, also known as MORF), which leads to continuous activation of PERK signaling pathway downstream of ER stress, and reduces the osteogenic differentiation ability of periodontal ligament stem cells (PDLSCs).
Subsequently, Li et al. [127] used low-intensity pulse ultrasound to up-regulate the osteogenic effect of PDLSCs under inflammatory conditions through UPR. Zhao et al. [128] demonstrated that JNK pathway activated by ER stress mediates TNF-ɑ-induced inflammation in BMSCs. These studies have confirmed that inhibiting ER stress can effectively reduce inflammatory response and enhance the osteogenic differentiation ability of stem cells, which may provide new insights for improving stem cell osteogenic differentiation and treating inflammatory bone diseases such as osteoporosis, so that inhibiting ER stress under inflammatory conditions to promote osteogenesis has great potential.
Exosomes regulate osteogenesis by activating ER stress pathways
Studies have shown that miRNA from exosomes of different cellular origins can enter recipient cells and then regulate the expression of genes associated with osteogenesis at the translational level to regulate osteogenesis [129]. We have also reviewed the use of exosome-derived non-coding RNAs for osteogenesis before [130]. However, whether exosomes promote osteogenesis by regulating ER stress is unclear. Platelet-rich plasma (PRP) has been widely used in clinical repair of bone and soft tissue injuries. Recent studies have shown that PRP contains a large number of extracellular vesicles [131]. Tao et al. [132] found that PRP-derived exosomes (PRP-Exos) binds to related receptors, promotes Akt phosphorylation, activates β-catenin to promote osteogenesis, and activates Bcl-2 to inhibit GC-induced apoptosis and ER stress (Fig. 2). Exosomes show great potential in PRP repair tissues, which is closely related to downstream ER stress pathways. Wang et al. [133] reported that miR-485–5p modified exosomes inhibit ER stress and alleviate chondrocyte apoptosis for the treatment of OA. Liao et al. [134] demonstrated that BMSCs-derived exosomes (BMSCs-Exos) can improve the apoptosis of nucleus pulposus cells induced by ER stress. Can BMSCs-Exos attenuate osteoblast apoptosis by inhibiting excessive ER stress? This may be a new mechanism of exosome promoting osteogenesis, which needs to be verified by future experiments.

Copyright© The Authors 2017. Published by Ivyspring International Publisher
PRP-Exos rescued cells from GC-induced apoptosis via the Akt/Bcl-2 pathway [132]. Tao SC, Yuan T, Rui BY, Zhu ZZ, Guo SC, Zhang CQ. Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway. Theranostics. 2017;7(3):733–50.
Effects of ER stress pathways on angiogenesis
Appropriate ER stress contributes to angiogenesis
ER stress promotes the differentiation of monocytes into ECs, leading to angiogenesis [147, 148], suggesting that ER stress can promote angiogenesis. The angiogenic effect of ER stress is mediated by regulation of angiogenic factors by the UPR. Appropriate ER stress triggers the production of angiogenic factors [149, 150]; however, the mechanism is unclear.
Three UPR signaling pathways bind to regulatory regions of VEGFA, and jointly drive VEGFA transcription [151, 152]. ER stress initiates angiogenesis signaling via UPR-mediated upregulation of VEGFA [153, 154]. The inducible ER chaperone oxygen-regulated protein 150 (OPR150) promotes the expression of VEGFA in pathological conditions and is a potential target for regulating angiogenesis [155]. Under ER stress, the IRE1ɑ-XBP1 pathway promotes tumor angiogenesis [156,157,]– [158], the PERK-ATF4 pathway promotes bone angiogenesis [153, 159], and the ATF6 pathway promotes embryonic angiogenesis [160], by upregulating VEGFA. Binet et al. [161] reported a pro-angiogenic role for the UPR in diseases characterized by pathological vascular abnormalities. Therefore, targeted regulation of angiogenesis through the UPR has therapeutic potential for vascular necrotizing diseases. VEGFA can spontaneously increase in acute myocardial ischemia, inducing intracellular Ca2+ overload and activating ER stress in a positive-feedback loop [162]. Excessive ER stress induces BMSCs apoptosis [128], and VEGFA stimulates the differentiation of BMSCs into ECs, thus protecting BMSCs and promoting angiogenesis [163]. Increased spontaneous VEGFA production also promotes compensatory angiogenesis through the ROS-ER stress-autophagy axis [162].
The UPR also regulates vascular growth factors. For example, ER stress promotes angiogenesis by upregulating interleukin 8 (IL-8) [164], FGF2 [36], placental growth factor (PIGF) [165], and granulocyte–macrophage colony stimulating factor (GM-CSF) [166] via different transcriptional mechanisms. However, pentraxin 3 (PTX3) has a high affinity for FGF2 and can inhibit its angiogenesis [167, 168], but Ma et al. [169] found that the ATF4 pathway activates SMAD-specific E3 ubiquitin ligase 2 and leads to PTX3 degradation, thus promoting angiogenesis. Philippe et al. [36] demonstrated that the PERK pathway activates the translation of dependent internal ribosome entry site (IRES), thereby promoting the expression of the angiogenic factors VEGFA and FGF2. These studies have suggested potential therapeutic targets for ischemia in vascular necrotizing diseases.
Excessive ER stress impairs angiogenesis
Excessive ER stress impairs angiogenesis not only by reducing the transcription of pro-angiogenetic growth factors such as VEGFA [170,171,– 172] and PIGF [173] but also by activating negative angiogenic regulators such as delta-like 4 (DLL4) IRES [174]. Excessive ER stress can induce apoptosis of ECs [175,176,177,178], thus suppressing their angiogenesis [171]. Maamoun et al. [179] showed that ER stress causes EC dysfunction, suggesting that targeting ER stress could promote angiogenesis (Table 2). The ER stress-mediated decreased expression of angiogenic genes is related to age [180].
The anti-angiogenic effect of ER stress also has benefits, such as inhibiting cancer progression [181]. ER stress can induce the expression of miR-153, which inhibits angiogenesis by two mechanisms, suggesting a novel therapeutic strategy for breast cancer [182].
Regulation of ER stress pathways to interfere with angiogenesis
HG impairs angiogenesis by activating ER stress pathways
In diabetic retinopathy (DR), HG damages normal blood vessels and causes abnormal neovascularization [183, 184]. ER stress is closely related to retinal angiogenesis [185]. Wang et al. [186] showed that regulation of ER stress can inhibit abnormal neovascularization. However, whether damaged normal blood vessels can be restored by regulating ER stress is unknown.
HG rapidly activates ER stress in ECs [187] and angiogenic progenitor cells (APCs) [188], leading to microvascular EC dysfunction and impair angiogenesis [189]. ECs have a greater apoptotic effect under GC induction than do other cells [190, 191]. Gao et al. [116] demonstrated that GCs induce EC apoptosis by activating ER stress, leading to microvascular damage. Alleviating the ER stress induced by HG can counteract HG-induced EC apoptosis [172], thus restoring angiogenesis [192] and enhancing vascular repair by circulating angiogenic cells (CACs) [193] (Table 2). Inhibition of ER stress can prevent vascular damage by upregulating pro-angiogenic factors and downregulating anti-angiogenic factors [194]. Wang et al. [195] found that an atypical UPR pathway mediated by IRE1ɑ regulates miRs, thereby protecting the pro-angiogenic growth factor angiopoietin 1 (ANGPT1) from miR attack under HG conditions and promoting bone marrow–derived progenitor cell (BMPC) angiogenesis. Therefore, targeting ER stress is the key to reversing HG-induced vascular injury.
Regulation of ER stress pathways on angiogenesis under inflammatory conditions
In recent years, ER stress pathways secondary to inflammation have become new targets for intracellular therapy. ER stress can induce nucleotide-binding domain and leucine-rich repeat containing (NLRP3) inflammasome through PERK and IRElα pathways, regulate the release of inflammatory cytokines, and trigger inflammatory response [196]. Wang et al. [197] demonstrated that there is a positive feedback loop between interleukin-17A (IL-17A) and ER stress, and that inhibition of ER stress or IL-17A can reduce the neovascularization area of DR. At present, inhibition of ER stress can alleviate inflammation and inhibit angiogenesis, which has been proved in both cell and animal experiments [186, 198]. Although ER stress pathway shows great potential in anti-inflammatory and anti-vascular therapy, more in-depth mechanism studies are needed before clinical trials.
Exosomes regulate angiogenesis by activating ER stress pathways
Exosomes promote angiogenesis by inducing the regeneration of damaged blood vessels by inhibiting EC apoptosis and promoting their angiogenic activity [199,200,201,202]. Tumor cell-derived exosomes deliver miR-25-3p to ECs, thereby disrupting ECs integrity, increasing vascular permeability, and promoting angiogenesis, thereby promoting tumor metastasis [203]. Based on the role of ER stress in numerous pathological conditions, whether exosomes promote angiogenesis by regulating ER stress is a topic of interest. Tao et al. [132] have found that PRP-Exos activates the Akt pathway under ER stress, releasing multiple growth factors and promoting angiogenesis (Fig. 2).
Exosomes have a dual regulatory effect on angiogenesis. Angiogenesis can be inhibited by exosomes. For example, exosomal circular RNAs (circRNAs) act as signal carriers to trigger EC dysfunction [204], exosomes can enhance the inhibitory effect of the anti-angiogenic peptide KV11 on pathological retinal angiogenesis [205], and circulating exosomal miR-20b-5p is transferred to vascular ECs to inhibit the regeneration of diabetic damaged blood vessels [206]. Wang et al. showed that ER-stressed HN4 cell-derived exosomes modified by miR-424–5p inhibit angiogenesis by HUVECs [207].
Exosomes from different sources have different regulatory effects on angiogenesis under ER stress. Until now, studies on exosomes promoting angiogenesis by activating ER stress have focused on exosomes of tumor cell origin. Lin et al. [208] demonstrated that after knocking down PERK in HUVEC, HeLa cell-derived exosomes can significantly improve HUVEC proliferation. We know that BMSCs-Exos have great potential in promoting angiogenesis [209], but whether ER stress may be a downstream pathway and whether we can enhance the ability of BMSCs-Exos to promote angiogenesis by regulating ER stress needs to be demonstrated in future studies.
Potential interventions related to ER stress pathways
Because the ER controls protein synthesis and degradation, ER stress is used clinically to restore myogenic differentiation to treat uremic sarcopenia [215]. Moreover, clinical trials by Bella et al. [216] suggested that ER stress may play a key role in the pathogenesis of amyotrophic lateral sclerosis by altering the regulation of protein balance, and that molecules acting on functional control of the UPR pathway may be beneficial in slowing disease progression, but subgroup analyses were not performed in this study. Therefore, this effect on targeting ER stress is considered exploratory. Besides, drugs targeting the IRE1ɑ-XBP1 pathway can inhibit vascular smooth muscle apoptosis, thereby alleviating aortic dissection [217]. Dexmedetomidine pretreatment can effectively protect myocardial ischemia–reperfusion-induced acute kidney injury by inhibiting ER stress [218].
Regulation of ER-related signaling pathways is most commonly used in the treatment of tumor diseases. ER stress is an essential intermediate targeting pathway in tumor therapy. Activation of ER stress can increase the cytotoxicity of photodynamic therapy to tumor cells [219]. Chemotherapy can increase tumor (sarcoma and gastric cancer) sensitivity by activating ER stress [220, 221]. Use of some chemotherapy drugs is limited by their toxicity. However, drugs that inhibit ER stress have reduced toxicity, and can be used in chemotherapy for cancer [222, 223]. Basic research by Varone et al. [224] showed that ISRIB (a small molecule that inhibits the action of phosphorylated eIF2ɑ) increases ER protein load, reactivates protein synthesis in damaged protein homeostasis, and ultimately promotes tumor cytotoxicity. ISRIB offers a new treatment option that can effectively inhibit tumor progression in conditions with impaired protein balance.
Although the mechanism of ER stress has been relatively clear, the current research on the intervention effect of ER stress in many diseases such as different types of diabetes and its complications is far from enough. Regulating a key signaling pathway node in the complex process of ER stress to affect the occurrence and development of diseases is an important target for drug therapy exploration, which has important clinical guiding value and practical significance. Further large-scale and long-term studies are needed to confirm the clinical benefits of this new pharmacological protocol, which may provide a promising therapeutic approach for targeted therapies for a number of diseases in the clinic.
Conclusion and perspective
In regenerative medicine, bone defects can be improved by promoting angiogenesis and osteogenesis. Our research has focused on inducing the regeneration of dead blood vessels and bone. ER stress is involved in many diseases. ER stress is a double-edged sword; its activation can promote cell generation, but excessive activation can induce apoptosis. ER stress plays a dual role in osteogenesis and angiogenesis, and thereby determines cell fate. Here we systematically reviewed the effect of ER stress on osteogenesis and angiogenesis. ER stress can be activated in pathological conditions such as HG and inflammation, or by inducers, and is inactivated by inhibitors. Therefore, regulation of ER stress has potential as a therapeutic target to promote osteogenesis and angiogenesis. Although regulating ER stress stimulates osteogenesis and angiogenesis, the mechanism is unclear. Efforts should focus on unraveling the mechanisms underlying the roles of ER stress in osteogenesis and angiogenesis.
Acellular therapy, such as exosome-mediated regulation of ER stress, is a focus of research. BMSCs-Exos have great potential for osteogenesis and angiogenesis, and we propose to hypothesize that ER stress can act as a downstream pathway for their regulation. Our future studies will further clarify the mechanism by which BMSCs-Exos promote angiogenesis and bone regeneration by regulating ER stress. Further research on the mechanism of ER stress regulating osteogenesis and angiogenesis will be helpful for the repair of bone defects.
Availability of data and materials
Not applicable.
Abbreviations
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- HG:
-
High glucose
- ECs:
-
Endothelial cells
- VEGF:
-
Vascular endothelial growth factor
- FGF:
-
Fibroblast growth factor
- ERQC:
-
ER quality control
- GPR:
-
Glucose-regulated protein
- PERK:
-
Protein kinase-R-like ER kinase
- IRE1ɑ:
-
Inositol-requiring enzyme 1ɑ
- ATF6:
-
Activating transcription factor 6
- BiP:
-
Immunoglobulin heavy-chain binding protein
- GPR78:
-
G protein coupled receptor 78
- XBP1:
-
X-box binding protein
- eIF2ɑ:
-
Eukaryotic initiation factor 2ɑ
- PDI:
-
Protein disulfide isomerase
- ATF6p90:
-
Full-length ATF6
- S1P:
-
Site 1 protease
- S2P:
-
Site 2 protease
- ATF6p50:
-
50 KDa fragment
- CHOP:
-
CCAAT-enhancer-binding protein homologus protein
- ASK1:
-
Apoptosis signal regulating kinase 1
- JNK:
-
C-jun kinase
- GADD153:
-
Growth arrest and DNA damage-inducible 153
- ROS:
-
Reactive oxygen species
- Ca2 + :
-
Calcium
- Bcl-2:
-
B-cell lymphoma 2
- RIDD:
-
Regulated IRE1ɑ-dependent decay
- TRAF:
-
Tumor necrosis factor receptor associated factor
- ERAD:
-
ER-associated protein degradation
- PP1:
-
Protein phosphatase 1
- CreP:
-
Constitutive repressor of eIF2ɑ phosphorylation
- DR5:
-
Death receptor 5
- TXNIP:
-
Thioredoxin-interacting protein
- IP3R:
-
Inositol-1,4,5-triphosphate receptor
- BI-1:
-
Bax inhibitor-1
- GADD34:
-
Growth arrest and DNA damage inducible gene 34
- OA:
-
Osteoarthritis
- IDD:
-
Intervertebral disc degeneration
- SCI:
-
Spinal cord injury
- SAR1A:
-
Secretion-associated Ras-related GTPase 1A
- OI:
-
Osteogenesis imperfecta
- BMPs:
-
Bone morphogenetic proteins
- BMSCs:
-
Bone marrow mesenchymal stem cells
- Osx:
-
Osterix
- GCs:
-
Glucocorticoids
- ONFH:
-
Osteonecrosis of the femoral head
- GEN:
-
Geniposide
- 4-PBA:
-
4-Phenylbutyric acid
- PRP-Exos:
-
Exosomes derived from platelet-rich plasma
- TNF-ɑ:
-
Tumor necrosis factor-ɑ
- METTL3:
-
Methyltransferase-like 3
- CDs:
-
Carbon dots
- PI:
-
Proteasome inhibitor
- AGE:
-
Advanced glycation end product
- PDLSC:
-
Periodontal ligament stem cell
- MNT:
-
Micro-/nano-topography
- PA:
-
Palmitate
- FTO:
-
Fat mass and obesity associated
- OPR150:
-
Oxygen-regulated protein 150
- IL-8:
-
Interleukin 8
References
Bharadwaz A, Jayasuriya AC. Osteogenic differentiation cues of the bone morphogenetic protein-9 (BMP-9) and its recent advances in bone tissue regeneration. Mater Sci Eng C Mater Biol Appl. 2021;120: 111748.
Yuan B, Wang L, Zhao R, Yang X, Yang X, Zhu X, Liu L, Zhang K, Song Y, Zhang X. A biomimetically hierarchical polyetherketoneketone scaffold for osteoporotic bone repair. Sci Adv. 2020;6:eabc4704.
Jiao H, **ao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. 2015;13:327–35.
Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, Boemi M, Giugliano D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. 2008;57:1349–54.
Cai F, Liu Y, Liu K, Zhao R, Chen W, Yusufu A, Liu Y. Diabetes mellitus impairs bone regeneration and biomechanics. J Orthop Surg Res. 2023;18:169.
Sun X, Ma Z, Zhao X, ** W, Zhang C, Ma J, Qiang L, Wang W, Deng Q, Yang H, Zhao J, Liang Q, Zhou X, Li T, Wang J. Three-dimensional bioprinting of multicell-laden scaffolds containing bone morphogenic protein-4 for promoting M2 macrophage polarization and accelerating bone defect repair in diabetes mellitus. Bioact Mater. 2021;6:757–69.
Wu Z, Bai J, Ge G, Wang T, Feng S, Ma Q, Liang X, Li W, Zhang W, Xu Y, Guo K, Cui W, Zha G, Geng D. Regulating macrophage polarization in high glucose microenvironment using lithium-modified bioglass-hydrogel for diabetic bone regeneration. Adv Healthc Mater. 2022;11: e2200298.
Zhang D, Wu Y, Li Z, Chen H, Huang S, Jian C, Yu A. MiR-144-5p, an exosomal miRNA from bone marrow-derived macrophage in type 2 diabetes, impairs bone fracture healing via targeting Smad1. J Nanobiotechnology. 2021;19:226.
Rather HA, Jhala D, Vasita R. Dual functional approaches for osteogenesis coupled angiogenesis in bone tissue engineering. Mater Sci Eng C Mater Biol Appl. 2019;103: 109761.
Schott NG, Friend NE, Stegemann JP. Coupling osteogenesis and vasculogenesis in engineered orthopedic tissues. Tissue Eng Part B Rev. 2021;27:199–214.
Grellier M, Bordenave L, Amedee J. Cell-to-cell communication between osteogenic and endothelial lineages: implications for tissue engineering. Trends Biotechnol. 2009;27:562–71.
Qin Y, Sun R, Wu C, Wang L, Zhang C. Exosome: a novel approach to stimulate bone regeneration through regulation of osteogenesis and angiogenesis. Int J Mol Sci. 2016;17:712.
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–8.
Huang Z, Bao SD. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J Gastroenterol. 2004;10:463–70.
Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984;223:1296–9.
Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–94.
Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–94.
Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov. 2022;21:115–40.
Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9.
Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23:143–9.
van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–705.
Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, Matsuhisa K, Saito A, Imaizumi K. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun. 2016;480:166–72.
Gurunathan S, Kim JH. Graphene oxide enhances biogenesis and release of exosomes in human ovarian cancer cells. Int J Nanomedicine. 2022;17:5697–731.
Gurunathan S, Kang MH, Jeyaraj M, Kim JH. Palladium nanoparticle-induced oxidative stress, endoplasmic reticulum stress, apoptosis, and immunomodulation enhance the biogenesis and release of exosome in human leukemia monocytic cells (THP-1). Int J Nanomedicine. 2021;16:2849–77.
Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J. 2008;27:315–27.
Romero-Brey I, Bartenschlager R. Endoplasmic reticulum: the favorite intracellular niche for viral replication and assembly. Viruses. 2016;8:160.
Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–90.
Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy–cooperation and control. Autophagy. 2015;11:200–13.
Shin YJ, Vavra U, Strasser R. Proper protein folding in the endoplasmic reticulum is required for attachment of a glycosylphosphatidylinositol anchor in plants. Plant Physiol. 2021;186:1878–92.
Wiseman RL, Mesgarzadeh JS, Hendershot LM. Resha** endoplasmic reticulum quality control through the unfolded protein response. Mol Cell. 2022;82:1477–91.
Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68–69:355–65.
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90.
Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999;79:683–701.
Hu H, Tian M, Ding C, Yu S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083.
Liu D, Zhang Y, Li X, Li J, Yang S, **ng X, Fan G, Yokota H, Zhang P. eIF2alpha signaling regulates ischemic osteonecrosis through endoplasmic reticulum stress. Sci Rep. 2017;7:5062.
Philippe C, Dubrac A, Quelen C, Desquesnes A, Van Den Berghe L, Segura C, Filleron T, Pyronnet S, Prats H, Brousset P, Touriol C. PERK mediates the IRES-dependent translational activation of mRNAs encoding angiogenic growth factors after ischemic stress. Sci Signal. 2016;9:ra44.
Wang Y, Gao L, Li Y, Chen H, Sun Z. Nifedipine protects INS-1 beta-cell from high glucose-induced ER stress and apoptosis. Int J Mol Sci. 2011;12:7569–80.
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–38.
Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71–88.
Shiu RP, Pouyssegur J, Pastan I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin Rous sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci USA. 1977;74:3840–4.
Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J Biol Chem. 1989;264:20602–7.
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–4.
Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206.
Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404.
Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867-1882 e1821.
Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chavez-Arroyo A, Tsai AY, Cevallos SA, Winter MG, Pham OH, Tiffany CR, de Jong MF, Kerrinnes T, Ravindran R, Luciw PA, McSorley SJ, Baumler AJ, Tsolis RM. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394–7.
Preissler S, Ron D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol. 2019;11: a033894.
Behnke J, Feige MJ, Hendershot LM. BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J Mol Biol. 2015;427:1589–608.
Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: the Achilles heel of pancreatic beta cells. Mol Metab. 2017;6:1024–39.
Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic reticulum stress and the hallmarks of cancer, trends. Cancer. 2016;2:252–62.
Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69:927–47.
Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, Schell M, Sandoval-Espinola WJ, Tao J, Sha B, Graham M, Crooke R, Kleinridders A, Balskus EP, Rey FE, Morris AJ, Biddinger SB. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019;30:1141-1151.e1145.
Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–38.
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, Wang D, **ng J, Hou B, Li H, ** H, Du W, Zou Z. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38:402.
Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, de Mingo Pulido A, Mandula J, Zhang S, Thevenot P, Biswas S, Abdalla SK, Costich TL, Hanggi K, Anadon CM, Flores ER, Haura EB, Mehrotra S, Pilon-Thomas S, Ruffell B, Munn DH, Cubillos-Ruiz JR, Conejo-Garcia JR, Rodriguez PC. The unfolded protein response mediator pERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. 2020;52:668-682 e667.
Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692–706.
Jheng JR, Lau KS, Lan YW, Horng JT. A novel role of ER stress signal transducer ATF6 in regulating enterovirus A71 viral protein stability. J Biomed Sci. 2018;25:9.
Hillary RF, FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. J Biomed Sci. 2018;25:48.
Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–76.
Hwang J, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci. 2018;43:593–605.
Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–81.
Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22:946–58.
Zuo S, Kong D, Wang C, Liu J, Wang Y, Wan Q, Yan S, Zhang J, Tang J, Zhang Q, Lyu L, Li X, Shan Z, Qian L, Shen Y, Yu Y. CRTH2 promotes endoplasmic reticulum stress-induced cardiomyocyte apoptosis through m-calpain. EMBO Mol Med. 2018;10: e8237.
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9.
Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–502.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59.
Liu Y, Zhu H, Yan X, Gu H, Gu Z, Liu F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem Biophys Res Commun. 2017;491:368–73.
Zhu Z, Gao S, Chen C, Xu W, **ao P, Chen Z, Du C, Chen B, Gao Y, Wang C, Liao J, Huang W. The natural product salicin alleviates osteoarthritis progression by binding to IRE1alpha and inhibiting endoplasmic reticulum stress through the IRE1alpha-IkappaBalpha-p65 signaling pathway. Exp Mol Med. 2022;54:1927–39.
Sim HJ, Cho C, Kim HE, Hong JY, Song EK, Kwon KY, Jang DG, Kim SJ, Lee HS, Lee C, Kwon T, Yang S, Park TJ. Augmented ERAD (ER-associated degradation) activity in chondrocytes is necessary for cartilage development and maintenance. Sci Adv. 2022;8:eabl4222.
Li Y, Zhang J, Zhou K, **e L, **ang G, Fang M, Han W, Wang X, **ao J. Elevating sestrin2 attenuates endoplasmic reticulum stress and improves functional recovery through autophagy activation after spinal cord injury. Cell Biol Toxicol. 2021;37:401–19.
Huang Z, Gong J, Lin W, Feng Z, Ma Y, Tu Y, Cai X, Liu J, Lv C, Lv X, Wu Q, Lu W, Zhao J, Ying Y, Li S, Ni W, Chen H. Catalpol as a component of Rehmannia glutinosa protects spinal cord injury by inhibiting endoplasmic reticulum stress-mediated neuronal apoptosis. Front Pharmacol. 2022;13: 860757.
Zhan F, Deng Q, Chen Z, **e C, **ang S, Qiu S, Tian L, Wu C, Ou Y, Chen J, Xu L. SAR1A regulates the RhoA/YAP and autophagy signaling pathways to influence osteosarcoma invasion and metastasis. Cancer Sci. 2022. https://doi.org/10.1111/cas.15551.
Scheiber AL, Guess AJ, Kaito T, Abzug JM, Enomoto-Iwamoto M, Leikin S, Iwamoto M, Otsuru S. Endoplasmic reticulum stress is induced in growth plate hypertrophic chondrocytes in G610C mouse model of osteogenesis imperfecta. Biochem Biophys Res Commun. 2019;509:235–40.
Duran I, Zieba J, Csukasi F, Martin JH, Wachtell D, Barad M, Dawson B, Fafilek B, Jacobsen CM, Ambrose CG, Cohn DH, Krejci P, Lee BH, Krakow D. 4-PBA treatment improves bone phenotypes in the Aga2 mouse model of osteogenesis imperfecta. J Bone Miner Res. 2022;37:675–86.
**ang Z, Wu Q, Wang Y, Wang P, He Y, Li J. eIF2alpha-ATF4 pathway activated by a change in the calcium environment participates in BCP-mediated bone regeneration. ACS Biomater Sci Eng. 2021;7:3256–68.
Zheng C, Attarilar S, Li K, Wang C, Liu J, Wang L, Yang J, Tang Y. 3D-printed HA15-loaded β-tricalcium phosphate/poly (lactic-co-glycolic acid) bone tissue scaffold promotes bone regeneration in rabbit radial defects. Int J Bioprinting. 2021;7:317.
Strong AL, Spreadborough PJ, Dey D, Yang P, Li S, Lee A, Haskins RM, Grimm PD, Kumar R, Bradley MJ, Yu PB, Levi B, Davis TA. BMP ligand trap ALK3-Fc attenuates osteogenesis and heterotopic ossification in blast-related lower extremity trauma. Stem Cells Dev. 2021;30:91–105.
Chen L, Zou X, Zhang RX, Pi CJ, Wu N, Yin LJ, Deng ZL. IGF1 potentiates BMP9-induced osteogenic differentiation in mesenchymal stem cells through the enhancement of BMP/Smad signaling. BMB Rep. 2016;49:122–7.
Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–83.
Zhang J, Weng Y, Liu X, Wang J, Zhang W, Kim SH, Zhang H, Li R, Kong Y, Chen X, Shui W, Wang N, Zhao C, Wu N, He Y, Nan G, Chen X, Wen S, Zhang H, Deng F, Wan L, Luu HH, Haydon RC, Shi LL, He TC, Shi Q. Endoplasmic reticulum (ER) stress inducible factor cysteine-rich with EGF-like domains 2 (Creld2) is an important mediator of BMP9-regulated osteogenic differentiation of mesenchymal stem cells. PLoS ONE. 2013;8: e73086.
Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia. 2019;33:918–30.
Horiuchi K, Tohmonda T, Morioka H. The unfolded protein response in skeletal development and homeostasis. Cell Mol Life Sci. 2016;73:2851–69.
Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; contributing factors to apoptosis and osteoarthritis. Int J Mol Sci. 2017;18:665.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29.
Kousteni S. Osterix finds its master. EMBO Rep. 2011;12:382–3.
Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, Morioka H, Nakamura M, Iwawaki T, Chiba K, Toyama Y, Urano F, Horiuchi K. The IRE1alpha-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011;12:451–7.
He S, Fu T, Yu Y, Liang Q, Li L, Liu J, Zhang X, Zhou Q, Guo Q, Xu D, Chen Y, Wang X, Chen Y, Liu J, Gan Z, Liu Y. IRE1alpha regulates skeletal muscle regeneration through myostatin mRNA decay. J Clin Invest. 2021. https://doi.org/10.1172/JCI143737.
Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. J Atheroscler Thromb. 2018;25:294–303.
Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry syndrome. Cell. 2004;117:387–98.
Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809–18.
Shi M, Song W, Han T, Chang B, Li G, ** J, Zhang Y. Role of the unfolded protein response in topography-induced osteogenic differentiation in rat bone marrow mesenchymal stem cells. Acta Biomater. 2017;54:175–85.
Yang SY, Wei FL, Hu LH, Wang CL. PERK-eIF2alpha-ATF4 pathway mediated by endoplasmic reticulum stress response is involved in osteodifferentiation of human periodontal ligament cells under cyclic mechanical force. Cell Signal. 2016;28:880–6.
Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol. 2008;217:693–707.
Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB, Kim SH, Choi HS, Koh JT. BMP2 protein regulates osteocalcin expression via Runx2-mediated Atf6 gene transcription. J Biol Chem. 2012;287:905–15.
Park SJ, Kim KJ, Kim WU, Oh IH, Cho CS. Involvement of endoplasmic reticulum stress in homocysteine-induced apoptosis of osteoblastic cells. J Bone Miner Metab. 2012;30:474–84.
Xue P, Li B, An Y, Sun J, He X, Hou R, Dong G, Fei D, ** F, Wang Q, ** Y. Decreased MORF leads to prolonged endoplasmic reticulum stress in periodontitis-associated chronic inflammation. Cell Death Differ. 2016;23:1862–72.
Li H, Li D, Ma Z, Qian Z, Kang X, ** X, Li F, Wang X, Chen Q, Sun H, Wu S. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy. 2018;14:1726–41.
Pereira RC, Stadmeyer LE, Smith DL, Rydziel S, Canalis E. CCAAT/enhancer-binding protein homologous protein (CHOP) decreases bone formation and causes osteopenia. Bone. 2007;40:619–26.
Wu CT, Chen YW, Su YH, Chiu CY, Guan SS, Yang RS, Liu SH. Gender difference of CCAAT/enhancer binding protein homologous protein deficiency in susceptibility to osteopenia. J Orthop Res. 2019;37:942–7.
Shirakawa K, Maeda S, Gotoh T, Hayashi M, Shinomiya K, Ehata S, Nishimura R, Mori M, Onozaki K, Hayashi H, Uematsu S, Akira S, Ogata E, Miyazono K, Imamura T. CCAAT/enhancer-binding protein homologous protein (CHOP) regulates osteoblast differentiation. Mol Cell Biol. 2006;26:6105–16.
Yan Y, Wang G, Luo X, Zhang P, Peng S, Cheng X, Wang M, Yang X. Endoplasmic reticulum stress-related calcium imbalance plays an important role on Zinc oxide nanoparticles-induced failure of neural tube closure during embryogenesis. Environ Int. 2021;152: 106495.
Vassalle M, Lin C-I. Calcium overload and cardiac function. J Biomed Sci. 2004;11:542–65.
Mohsin AA, Thompson J, Hu Y, Hollander J, Lesnefsky EJ, Chen Q. Endoplasmic reticulum stress-induced complex I defect: central role of calcium overload. Arch Biochem Biophys. 2020;683: 108299.
Cheng Z, Liu Y, Ma M, Sun S, Ma Z, Wang Y, Yu L, Qian X, Sun L, Zhang X, Liu Y, Wang Y. Lansoprazole-induced osteoporosis via the IP3R- and SOCE-mediated calcium signaling pathways. Mol Med. 2022;28:21.
Cui C, Lin T, Gong Z, Zhu Y. Relationship between autophagy, apoptosis and endoplasmic reticulum stress induced by melatonin in osteoblasts by septin7 expression. Mol Med Rep. 2020;21:2427–34.
Liu W, Xu C, Ran D, Wang Y, Zhao H, Gu J, Liu X, Bian J, Yuan Y, Liu Z. CaMK mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology. 2018;406–407:70–80.
Li X, Meng L, Wang F, Hu X, Yu Y. Sodium fluoride induces apoptosis and autophagy via the endoplasmic reticulum stress pathway in MC3T3-E1 osteoblastic cells. Mol Cell Biochem. 2019;454:77–85.
Wang J, Zhao Y, Cheng X, Li Y, Xu H, Manthari RK, Wang J. Effects of different Ca(2+) level on fluoride-induced apoptosis pathway of endoplasmic reticulum in the rabbit osteoblast in vitro. Food Chem Toxicol. 2018;116:189–95.
Che J, Lv H, Yang J, Zhao B, Zhou S, Yu T, Shang P. Iron overload induces apoptosis of osteoblast cells via eliciting ER stress-mediated mitochondrial dysfunction and p-eIF2alpha/ATF4/CHOP pathway in vitro. Cell Signal. 2021;84: 110024.
Tan J, Zhou Y, Luo J, Wu X, Liu H, Wang W, Li Z, Zhong M, Wu L, Li X. High glucose inhibits the osteogenic differentiation of periodontal ligament stem cells in periodontitis by activating endoplasmic reticulum stress. Ann Transl Med. 2022;10:204.
Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. 2017;17:233–47.
Chang C, Greenspan A, Gershwin ME. The pathogenesis, diagnosis and clinical manifestations of steroid-induced osteonecrosis. J Autoimmun. 2020;110: 102460.
Liu W, Zhao Z, Na Y, Meng C, Wang J, Bai R. Dexamethasone-induced production of reactive oxygen species promotes apoptosis via endoplasmic reticulum stress and autophagy in MC3T3-E1 cells. Int J Mol Med. 2018;41:2028–36.
Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015;73:60–8.
Huang J, Ye Y, **ao Y, Ren Q, Zhou Q, Zhong M, Jiao L, Wu L. Geniposide ameliorates glucocorticoid-induced osteoblast apoptosis by activating autophagy. Biomed Pharmacother. 2022;155: 113829.
Gao Y, Zhu H, Wang Q, Feng Y, Zhang C. Inhibition of PERK signaling prevents against glucocorticoid-induced endotheliocyte apoptosis and osteonecrosis of the femoral head. Int J Biol Sci. 2020;16:543–52.
**ao Y, Ren Q, Zheng Y, Zhang S, Ouyang J, Jiao L, Tang C, Li L, Shi W, Wang M, Zhang S, Zhang D, Zhong B, Peng F, Chen Z, Wu L. Geniposide ameliorated dexamethasone-induced endoplasmic reticulum stress and mitochondrial apoptosis in osteoblasts. J Ethnopharmacol. 2022;291: 115154.
Feng Y, Zhang R, Wang YR, Chen F, Luo Q, Cai C, Jiao Y, Xue P. Inhibition of endoplasmic reticulum stress by 4-phenyl butyric acid presents therapeutic effects on periodontitis: experimental studies in vitro and in rats. Stem Cells Int. 2021;2021:6618943.
Zhou R, Ma Y, Tao Z, Qiu S, Gong Z, Tao L, Zhu Y. Melatonin inhibits glucose-induced apoptosis in osteoblastic cell line through PERK-eIF2alpha-ATF4 pathway. Front Pharmacol. 2020;11: 602307.
Yang L, Liu S, Mu S, Guo R, Zhou L, Fu Q. Paeoniflorin attenuates dexamethasone-induced apoptosis of osteoblast cells and promotes bone formation via regulating AKT/mTOR/autophagy signaling pathway. Evid Based Complement Alternat Med. 2021;2021:6623464.
Duangchan T, Tawonsawatruk T, Angsanuntsukh C, Trachoo O, Hongeng S, Kitiyanant N, Supokawej A. Amelioration of osteogenesis in iPSC-derived mesenchymal stem cells from osteogenesis imperfecta patients by endoplasmic reticulum stress inhibitor. Life Sci. 2021;278:119628.
Li J, Yang S, Li X, Liu D, Wang Z, Guo J, Tan N, Gao Z, Zhao X, Zhang J, Gou F, Yokota H, Zhang P. Role of endoplasmic reticulum stress in disuse osteoporosis. Bone. 2017;97:2–14.
Tang J, Wu T, **ong J, Su Y, Zhang C, Wang S, Tang Z, Liu Y. Porphyromonas gingivalis lipopolysaccharides regulate functions of bone marrow mesenchymal stem cells. Cell Prolif. 2015;48:239–48.
Kuang W, Zheng L, Xu X, Lin Y, Lin J, Wu J, Tan J. Dysregulation of the miR-146a-Smad4 axis impairs osteogenesis of bone mesenchymal stem cells under inflammation. Bone Res. 2017;5:17037.
Victor P, Sarada D, Ramkumar KM. Crosstalk between endoplasmic reticulum stress and oxidative stress: focus on protein disulfide isomerase and endoplasmic reticulum oxidase 1. Eur J Pharmacol. 2021;892: 173749.
** S, Park JY, Hong JM, Kim TH, Shin HI, Park EK, Kim SY. Inhibitory effect of (-)-epigallocatechin gallate on titanium particle-induced TNF-α release and in vivo osteolysis. Exp Mol Med. 2011;43:411–8.
Li H, Deng Y, Tan M, Feng G, Kuang Y, Li J, Song J. Low-intensity pulsed ultrasound upregulates osteogenesis under inflammatory conditions in periodontal ligament stem cells through unfolded protein response. Stem Cell Res Ther. 2020;11:215.
Zhao X, Zhang G, Wu L, Tang Y, Guo C. Inhibition of ER stress-activated JNK pathway attenuates TNF-alpha-induced inflammatory response in bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2021;541:8–14.
Ma S, Zhang Y, Li S, Li A, Li Y, Pei D. Engineering exosomes for bone defect repair. Front Bioeng Biotechnol. 2022;10:1091360.
Ren YZ, Ding SS, Jiang YP, Wen H, Li T. Application of exosome-derived noncoding RNAs in bone regeneration: opportunities and challenges. World J Stem Cells. 2022;14:473–89.
Qu M, Zou X, Fang F, Wang S, Xu L, Zeng Q, Fan Z, Chen L, Yue W, **e X, Pei X. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat Commun. 2020;11:4964.
Tao SC, Yuan T, Rui BY, Zhu ZZ, Guo SC, Zhang CQ. Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway. Theranostics. 2017;7:733–50.
Wang Y, Fan A, Lu L, Pan Z, Ma M, Luo S, Liu Z, Yang L, Cai J, Yin F. Exosome modification to better alleviates endoplasmic reticulum stress induced chondrocyte apoptosis and osteoarthritis. Biochem Pharmacol. 2022;206: 115343.
Liao Z, Luo R, Li G, Song Y, Zhan S, Zhao K, Hua W, Zhang Y, Wu X, Yang C. Exosomes from mesenchymal stem cells modulate endoplasmic reticulum stress to protect against nucleus pulposus cell death and ameliorate intervertebral disc degeneration in vivo. Theranostics. 2019;9:4084–100.
Son HE, Kim EJ, Jang WG. Curcumin induces osteoblast differentiation through mild-endoplasmic reticulum stress-mediated such as BMP2 on osteoblast cells. Life Sci. 2018;193:34–9.
Kong Y, Zhang Y, Cai Y, Li D, Yi B, Xu Q. METTL3 mediates osteoblast apoptosis by regulating endoplasmic reticulum stress during LPS-induced inflammation. Cell Signal. 2022;95: 110335.
** N, ** N, Wang Z, Liu L, Meng L, Li D, Li X, Zhou D, Liu J, Bu W, Sun H, Yang B. Osteopromotive carbon dots promote bone regeneration through the PERK-eIF2alpha-ATF4 pathway. Biomater Sci. 2020;8:2840–52.
Zhang D, De Veirman K, Fan R, Jian Q, Zhang Y, Lei L, Evans H, Wang Y, Lei L, Wang B, Williamson RA, Chantry A, He P, Li A, De Raeve H, Vanderkerken K, He A, Hu J. ER stress arm XBP1s plays a pivotal role in proteasome inhibition-induced bone formation. Stem Cell Res Ther. 2020;11:516.
Meng X, Zhu Y, Tao L, Zhao S, Qiu S. Periostin has a protective role in melatonininduced cell apoptosis by inhibiting the eIF2alphaATF4 pathway in human osteoblasts. Int J Mol Med. 2018;41:1003–12.
Suzuki R, Fujiwara Y, Saito M, Arakawa S, Shirakawa JI, Yamanaka M, Komohara Y, Marumo K, Nagai R. Intracellular accumulation of advanced glycation end products induces osteoblast apoptosis via endoplasmic reticulum stress. J Bone Miner Res. 2020;35:1992–2003.
Tan J, Zhou L, Xue P, An Y, Luo L, Zhang R, Wu G, Wang Y, Zhu H, Wang Q. Tumor necrosis factor-alpha attenuates the osteogenic differentiation capacity of periodontal ligament stem cells by activating PERK signaling. J Periodontol. 2016;87:e159-171.
Han C, **e K, Yang C, Zhang F, Liang Q, Lan C, Chen J, Huang K, Liu J, Li K, Tang Y, Wang L. HA15 alleviates bone loss in ovariectomy-induced osteoporosis by targeting HSPA5. Exp Cell Res. 2021;406: 112781.
Mengqi S, Wen S, Boxin Z, Minni L, Yan Z, Qun W, Yumei Z. Micro/nano topography with altered nanotube diameter differentially trigger endoplasmic reticulum stress to mediate bone mesenchymal stem cell osteogenic differentiation. Biomed Mater. 2020;16: 015024.
**g L, Jia XW. Lycium barbarum polysaccharide arbitrates palmitate-induced apoptosis in MC3T3E1 cells through decreasing the activation of ERSmediated apoptosis pathway. Mol Med Rep. 2018;17:2415–21.
Son HE, Min HY, Kim EJ, Jang WG. Fat mass and obesity-associated (FTO) stimulates osteogenic differentiation of C3H10T1/2 cells by inducing mild endoplasmic reticulum stress via a positive feedback loop with p-AMPK. Mol Cells. 2020;43:58–65.
Yang F, Tang J, Dai K, Huang Y. Metallic wear debris collected from patients induces apoptosis in rat primary osteoblasts via reactive oxygen speciesmediated mitochondrial dysfunction and endoplasmic reticulum stress. Mol Med Rep. 2019;19:1629–37.
Niu J, Wang K, Zhelyabovska O, Saad Y, Kolattukudy PE. MCP-1-induced protein promotes endothelial-like and angiogenic properties in human bone marrow monocytic cells. J Pharmacol Exp Ther. 2013;347:288–97.
Roy A, Kolattukudy PE. Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell Signal. 2012;24:2123–31.
Soczewski E, Grasso E, Gallino L, Hauk V, Fernandez L, Gori S, Paparini D, Perez Leiros C, Ramhorst R. Immunoregulation of the decidualization program: focus on the endoplasmic reticulum stress. Reproduction. 2020;159:R203–11.
Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE. 2010;5: e12521.
Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE. 2010;5: e9575.
Miyagi H, Kanemoto S, Saito A, Asada R, Iwamoto H, Izumi S, Kido M, Gomi F, Nishida K, Kiuchi Y, Imaizumi K. Transcriptional regulation of VEGFA by the endoplasmic reticulum stress transducer OASIS in ARPE-19 cells. PLoS ONE. 2013;8: e55155.
Castranova D, Davis AE, Lo BD, Miller MF, Paukstelis PJ, Swift MR, Pham VN, Torres-Vazquez J, Bell K, Shaw KM, Kamei M, Weinstein BM. Aminoacyl-transfer RNA synthetase deficiency promotes angiogenesis via the unfolded protein response pathway. Arterioscler Thromb Vasc Biol. 2016;36:655–62.
Loeuillard E, El Mourabit H, Lei L, Lemoinne S, Housset C, Cadoret A. Endoplasmic reticulum stress induces inverse regulations of major functions in portal myofibroblasts during liver fibrosis progression. Biochim Biophys Acta Mol Basis Dis. 1864;2018:3688–96.
Ozawa K, Kondo T, Hori O, Kitao Y, Stern DM, Eisenmenger W, Ogawa S, Ohshima T. Expression of the oxygen-regulated protein ORP150 accelerates wound healing by modulating intracellular VEGF transport. J Clin Investig. 2001;108:41–50.
Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, Kaufman RJ, Chevet E, Bikfalvi A, Moenner M. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007;67:6700–7.
Wu SX, Ye SS, Hong YX, Chen Y, Wang B, Lin XJ, Lin X. Hepatitis B virus small envelope protein promotes hepatocellular carcinoma angiogenesis via endoplasmic reticulum stress signaling to upregulate the expression of vascular endothelial growth factor A. J Virol. 2022;96: e0197521.
Zeng L, **ao Q, Chen M, Margariti A, Martin D, Ivetic A, Xu H, Mason J, Wang W, Cockerill G, Mori K, Li JY, Chien S, Hu Y, Xu Q. Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation. 2013;127:1712–22.
Zhu K, Jiao H, Li S, Cao H, Galson DL, Zhao Z, Zhao X, Lai Y, Fan J, Im HJ, Chen D, **ao G. ATF4 promotes bone angiogenesis by increasing VEGF expression and release in the bone environment. J Bone Miner Res. 2013;28:1870–84.
Soczewski E, Gori S, Paparini D, Grasso E, Fernandez L, Gallino L, Schafir A, Irigoyen M, Lobo TF, Salamone G, Mattar R, Daher S, Perez Leiros C, Ramhorst R. VIP conditions human endometrial receptivity by privileging endoplasmic reticulum stress through ATF6alpha pathway. Mol Cell Endocrinol. 2020;516:110948.
Binet F, Sapieha P. ER stress and angiogenesis. Cell Metab. 2015;22:560–75.
Zou J, Fei Q, **ao H, Wang H, Liu K, Liu M, Zhang H, **ao X, Wang K, Wang N. VEGF-A promotes angiogenesis after acute myocardial infarction through increasing ROS production and enhancing ER stress-mediated autophagy. J Cell Physiol. 2019;234:17690–703.
Li J, Li Z, Wang C, Li Z, Xu H, Hu Y, Tan Z, Zhang F, Liu C, Yang M, Wang Y, ** Y, Peng Z, Biswas S, Zhu L. The regulatory effect of VEGF-Ax on rat bone marrow mesenchymal stem cells’ angioblastic differentiation and its proangiogenic ability. Stem Cells Dev. 2020;29:667–77.
Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA. 2006;103:12741–6.
Vandewynckel YP, Laukens D, Devisscher L, Bogaerts E, Paridaens A, Van den Bussche A, Raevens S, Verhelst X, Van Steenkiste C, Jonckx B, Libbrecht L, Geerts A, Carmeliet P, Van Vlierberghe H. Placental growth factor inhibition modulates the interplay between hypoxia and unfolded protein response in hepatocellular carcinoma. BMC Cancer. 2016;16:9.
Ying R, Li SW, Chen JY, Zhang HF, Yang Y, Gu ZJ, Chen YX, Wang JF. Endoplasmic reticulum stress in perivascular adipose tissue promotes destabilization of atherosclerotic plaque by regulating GM-CSF paracrine. J Transl Med. 2018;16:105.
O’Neill CL, Guduric-Fuchs J, Chambers SE, O’Doherty M, Bottazzi B, Stitt AW, Medina RJ. Endothelial cell-derived pentraxin 3 limits the vasoreparative therapeutic potential of circulating angiogenic cells. Cardiovasc Res. 2016;112:677–88.
Rusnati M, Camozzi M, Moroni E, Bottazzi B, Peri G, Indraccolo S, Amadori A, Mantovani A, Presta M. Selective recognition of fibroblast growth factor-2 by the long pentraxin PTX3 inhibits angiogenesis. Blood. 2004;104:92–9.
Ma M, Yang W, Cai Z, Wang P, Li H, Mi R, Jiang Y, **e Z, Sui P, Wu Y, Shen H. SMAD-specific E3 ubiquitin ligase 2 promotes angiogenesis by facilitating PTX3 degradation in MSCs from patients with ankylosing spondylitis. Stem Cells. 2021;39:581–99.
Yu H, Wen K, Zhou X, Zhang Y, Yan Z, Fu H, Zhu J, Zhu Y. Role of unfolded protein response in genital malformation/damage of male mice induced by flutamide. Hum Exp Toxicol. 2020;39:1690–9.
Abdelsalam S, Pasha M, El-Gamal H, Hasan M, Elrayess M, Zeidan A, Korashy H, Agouni A. Protein tyrosine phosphatase 1B inhibition improves endoplasmic reticulum stress-impaired endothelial cell angiogenic response: a critical role for cell survival. Mol Med Rep. 2021;24:1.
Maamoun H, Zachariah M, McVey JH, Green FR, Agouni A. Heme oxygenase (HO)-1 induction prevents endoplasmic reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem Pharmacol. 2017;127:46–59.
Lokeswara AW, Hiksas R, Irwinda R, Wibowo N. Preeclampsia: from cellular wellness to inappropriate cell death, and the roles of nutrition. Front Cell Dev Biol. 2021;9: 726513.
Jaud M, Philippe C, Van Den Berghe L, Segura C, Mazzolini L, Pyronnet S, Laurell H, Touriol C. The PERK Branch of the Unfolded Protein Response Promotes DLL4 Expression by Activating an Alternative Translation Mechanism. Cancers (Basel). 2019;11:142.
Tang V, Fu S, Rayner BS, Hawkins CL. 8-Chloroadenosine induces apoptosis in human coronary artery endothelial cells through the activation of the unfolded protein response. Redox Biol. 2019;26: 101274.
Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, **ao Q, Wang W, ** ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A. 2009;106:8326–31.
Huang J, Wan L, Lu H, Li X. High expression of active ATF6 aggravates endoplasmic reticulum stressinduced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol Med Rep. 2018;17:6483–9.
Zachariah M, Maamoun H, Milano L, Rayman MP, Meira LB, Agouni A. Endoplasmic reticulum stress and oxidative stress drive endothelial dysfunction induced by high selenium. J Cell Physiol. 2020;236:4348–59.
Maamoun H, Abdelsalam SS, Zeidan A, Korashy HM, Agouni A. Endoplasmic reticulum stress: a critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int J Mol Sci. 2019;20:1658.
Shimoda Y, Matsuo K, Ono K, Soma Y, Ueyama T, Matoba S, Yamada H, Ikeda K. Aging differentially alters the expression of angiogenic genes in a tissue-dependent manner. Biochem Biophys Res Commun. 2014;446:1243–9.
Bae H, Lee JY, Song G, Lim W. Fucosterol suppresses the progression of human ovarian cancer by inducing mitochondrial dysfunction and endoplasmic reticulum stress. Mar Drugs. 2020;18:261.
Liang H, **ao J, Zhou Z, Wu J, Ge F, Li Z, Zhang H, Sun J, Li F, Liu R, Chen C. Hypoxia induces miR-153 through the IRE1alpha-XBP1 pathway to fine tune the HIF1alpha/VEGFA axis in breast cancer angiogenesis. Oncogene. 2018;37:1961–75.
Madonna R, Giovannelli G, Confalone P, Renna FV, Geng YJ, De Caterina R. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: implications for diabetic retinopathy. Cardiovasc Diabetol. 2016;15:18.
Anitha RE, Janani R, Peethambaran D, Baskaran V. Lactucaxanthin protects retinal pigment epithelium from hyperglycemia-regulated hypoxia/ER stress/VEGF pathway mediated angiogenesis in ARPE-19 cell and rat model. Eur J Pharmacol. 2021;899: 174014.
Zhang SX, Ma JH, Bhatta M, Fliesler SJ, Wang JJ. The unfolded protein response in retinal vascular diseases: implications and therapeutic potential beyond protein folding. Prog Retin Eye Res. 2015;45:111–31.
Wang Y, Wang L, Guo H, Peng Y, Nie D, Mo J, Ye L. Knockdown of MALAT1 attenuates high-glucose-induced angiogenesis and inflammation via endoplasmic reticulum stress in human retinal vascular endothelial cells. Biomed Pharmacother. 2020;124: 109699.
Chen Y, Wang JJ, Li J, Hosoya KI, Ratan R, Townes T, Zhang SX. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia. 2012;55:2533–45.
Bhatta M, Ma JH, Wang JJ, Sakowski J, Zhang SX. Enhanced endoplasmic reticulum stress in bone marrow angiogenic progenitor cells in a mouse model of long-term experimental type 2 diabetes. Diabetologia. 2015;58:2181–90.
Lai DW, Lin KH, Sheu WH, Lee MR, Chen CY, Lee WJ, Hung YW, Shen CC, Chung TJ, Liu SH, Sheu ML. TPL2 (therapeutic targeting tumor progression locus-2)/ATF4 (activating transcription factor-4)/SDF1alpha (chemokine stromal cell-derived factor-alpha) axis suppresses diabetic retinopathy. Circ Res. 2017;121:e37–52.
El Zaoui I, Behar-Cohen F, Torriglia A. Glucocorticoids exert direct toxicity on microvasculature: analysis of cell death mechanisms. Toxicol Sci. 2015;143:441–53.
Yu QS, Guo WS, Cheng LM, Lu YF, Shen JY, Li P. Glucocorticoids significantly influence the transcriptome of bone microvascular endothelial cells of human femoral head. Chin Med J (Engl). 2015;128:1956–63.
Nie X, Tang W, Zhang Z, Yang C, Qian L, **e X, Qiang E, Zhao J, Zhao W, **ao L, Wang N. Procyanidin B2 mitigates endothelial endoplasmic reticulum stress through a PPARdelta-dependent mechanism. Redox Biol. 2020;37: 101728.
Bhatta M, Chatpar K, Hu Z, Wang JJ, Zhang SX. Reduction of endoplasmic reticulum stress improves angiogenic progenitor cell function in a mouse model of type 1 diabetes. Cell Death Dis. 2018;9:467.
Amin A, Choi SK, Galan M, Kassan M, Partyka M, Kadowitz P, Henrion D, Trebak M, Belmadani S, Matrougui K. Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J Pathol. 2012;227:165–74.
Wang JM, Qiu Y, Yang ZQ, Li L, Zhang K. Inositol-requiring enzyme 1 facilitates diabetic wound healing through modulating MicroRNAs. Diabetes. 2017;66:177–92.
Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, Yin XM, O’Riordan MX. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity. 2015;43:451–62.
Wang Y, Gao S, Gao S, Li N, **e B, Shen X. Blocking the interaction between interleukin-17A and endoplasmic reticulum stress in macrophage attenuates retinal neovascularization in oxygen-induced retinopathy. Cell Biosci. 2021;11:82.
Ozge G, Karaca U, Savran M, Usta G, Gulle K, Sevimli M, Cankara FN, Asci H. Salubrinal ameliorates inflammation and neovascularization via the caspase 3/Enos signaling in an alkaline-induced rat corneal neovascularization model. Medicina (Kaunas). 2023;59:323.
Chen L, Wang Y, Li S, Zuo B, Zhang X, Wang F, Sun D. Exosomes derived from GDNF-modified human adipose mesenchymal stem cells ameliorate peritubular capillary loss in tubulointerstitial fibrosis by activating the SIRT1/eNOS signaling pathway. Theranostics. 2020;10:9425–42.
Huang P, Wang L, Li Q, Tian X, Xu J, Xu J, **ong Y, Chen G, Qian H, ** C, Yu Y, Cheng K, Qian L, Yang Y. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2020;116:353–67.
Liao Z, Chen Y, Duan C, Zhu K, Huang R, Zhao H, Hintze M, Pu Q, Yuan Z, Lv L, Chen H, Lai B, Feng S, Qi X, Cai D. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics. 2021;11:268–91.
Olejarz W, Kubiak-Tomaszewska G, Chrzanowska A, Lorenc T. Exosomes in angiogenesis and anti-angiogenic therapy in cancers. Int J Mol Sci. 2020;21:5840.
Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, Zhou K, Liu X, Ren X, Wang F, Hu J, Zhu X, Yang W, Liao W, Li G, Ding Y, Liang L. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun. 2018;9:5395.
**ong L, Chen L, Wu L, He W, Chen D, Peng Z, Li J, Zhu X, Su L, Li Y, Gong Y, **ao H. Lipotoxicity-induced circGlis3 impairs beta cell function and is transmitted by exosomes to promote islet endothelial cell dysfunction. Diabetologia. 2022;65:188–205.
Dong X, Lei Y, Yu Z, Wang T, Liu Y, Han G, Zhang X, Li Y, Song Y, Xu H, Du M, Yin H, Wang X, Yan H. Exosome-mediated delivery of an anti-angiogenic peptide inhibits pathological retinal angiogenesis. Theranostics. 2021;11:5107–26.
**ong Y, Chen L, Yan C, Zhou W, Endo Y, Liu J, Hu L, Hu Y, Mi B, Liu G. Circulating Exosomal miR-20b-5p inhibition restores Wnt9b signaling and reverses diabetes-associated impaired wound healing. Small. 2020;16: e1904044.
Wang Z, Jiao P, Zhong Y, Ji H, Zhang Y, Song H, Du H, Ding X, Wu H. The endoplasmic reticulum-stressed head and neck squamous cell carcinoma cells induced exosomal miR-424-5p inhibits angiogenesis and migration of humanumbilical vein endothelial cells through LAMC1-mediated Wnt/beta-catenin signaling pathway. Cell Transplant. 2022;31:9636897221083548.
Lin Y, Zhang C, **ang P, Shen J, Sun W, Yu H. Exosomes derived from HeLa cells break down vascular integrity by triggering endoplasmic reticulum stress in endothelial cells. J Extracell Vesicles. 2020;9:1722385.
Nazari-Shafti TZ, Neuber S, Garcia Duran A, Xu Z, Beltsios E, Seifert M, Falk V, Stamm C. Human mesenchymal stromal cells and derived extracellular vesicles: Translational strategies to increase their proangiogenic potential for the treatment of cardiovascular disease. Stem Cells Transl Med. 2020;9:1558–69.
Lin D, Wu H, Zhou Z, Tao Z, Jia T, Gao W. Ginkgolide B improves multiterritory perforator flap survival by inhibiting endoplasmic reticulum stress and oxidative stress. J Invest Surg. 2021;34:610–6.
Li MT, Ke J, Guo SF, Wu Y, Bian YF, Shan LL, Liu QY, Huo YJ, Guo C, Liu MY, Liu YJ, Han Y. The protective effect of quercetin on endothelial cells injured by hypoxia and reoxygenation. Front Pharmacol. 2021;12: 732874.
Haas MJ, Jafri M, Wehmeier KR, Onstead-Haas LM, Mooradian AD. Inhibition of endoplasmic reticulum stress and oxidative stress by vitamin D in endothelial cells. Free Radic Biol Med. 2016;99:1–10.
Wang G, Nie JH, Bao Y, Yang X. Sulforaphane rescues ethanol-suppressed angiogenesis through oxidative and endoplasmic reticulum stress in chick embryos. J Agric Food Chem. 2018;66:9522–33.
Shangguan WJ, Zhang YH, Li ZC, Tang LM, Shao J, Li H. Naringin inhibits vascular endothelial cell apoptosis via endoplasmic reticulum stress and mitochondrialmediated pathways and promotes intraosseous angiogenesis in ovariectomized rats. Int J Mol Med. 2017;40:1741–9.
Jheng JR, Chen YS, Ao UI, Chan DC, Huang JW, Hung KY, Tarng DC, Chiang CK. The double-edged sword of endoplasmic reticulum stress in uremic sarcopenia through myogenesis perturbation. J Cachexia Sarcopenia Muscle. 2018;9:570–84.
Dalla Bella E, Bersano E, Antonini G, Borghero G, Capasso M, Caponnetto C, Chiò A, Corbo M, Filosto M, Giannini F, Spataro R, Lunetta C, Mandrioli J, Messina S, Monsurrò MR, Mora G, Riva N, Rizzi R, Siciliano G, Silani V, Simone I, Sorarù G, Tugnoli V, Verriello L, Volanti P, Furlan R, Nolan JM, Abgueguen E, Tramacere I, Lauria G. The unfolded protein response in amyotrophic later sclerosis: results of a phase 2 trial. Brain. 2021;144:2635–47.
Zhang W, Wang M, Gao K, Zhong X, **e Y, Dai L, Liu W, Liu Y, He X, Li S, Madhusudhan T, Wang H, Zeng H. Pharmacologic IRE1alpha kinase inhibition alleviates aortic dissection by decreasing vascular smooth muscle cells apoptosis. Int J Biol Sci. 2022;18:1053–64.
Tang C, Hu Y, Gao J, Jiang J, Shi S, Wang J, Geng Q, Liang X, Chai X. Dexmedetomidine pretreatment attenuates myocardial ischemia reperfusion induced acute kidney injury and endoplasmic reticulum stress in human and rat. Life Sci. 2020;257: 118004.
Szokalska A, Makowski M, Nowis D, Wilczynski GM, Kujawa M, Wojcik C, Mlynarczuk-Bialy I, Salwa P, Bil J, Janowska S, Agostinis P, Verfaillie T, Bugajski M, Gietka J, Issat T, Glodkowska E, Mrowka P, Stoklosa T, Hamblin MR, Mroz P, Jakobisiak M, Golab J. Proteasome inhibition potentiates antitumor effects of photodynamic therapy in mice through induction of endoplasmic reticulum stress and unfolded protein response. Cancer Res. 2009;69:4235–43.
Chen W, Zou P, Zhao Z, Chen X, Fan X, Vinothkumar R, Cui R, Wu F, Zhang Q, Liang G, Ji J. Synergistic antitumor activity of rapamycin and EF24 via increasing ROS for the treatment of gastric cancer. Redox Biol. 2016;10:78–89.
Chiu HW, Tseng YC, Hsu YH, Lin YF, Foo NP, Guo HR, Wang YJ. Arsenic trioxide induces programmed cell death through stimulation of ER stress and inhibition of the ubiquitin-proteasome system in human sarcoma cells. Cancer Lett. 2015;356:762–72.
Gao L, Yuan P, Wei Y, Fu Y, Hou Y, Li P, Chen Y, Ruan Y, Zhou N, Zheng X, Feng W. Total flavonoids of Selaginella tamariscina (P. Beauv.) Spring ameliorates doxorubicin-induced cardiotoxicity by modulating mitochondrial dysfunction and endoplasmic reticulum stress via activating MFN2/PERK. Phytomedicine. 2022;100: 154065.
Wang F, Han L. Upregulation of serum and glucocorticoid-regulated kinase 1 (SGK1) ameliorates doxorubicin-induced cardiotoxic injury, apoptosis, inflammation and oxidative stress by suppressing glucose regulated protein 78 (GRP78)-mediated endoplasmic reticulum stress. Bioengineered. 2022;13:844–55.
Varone E, Decio A, Barbera MC, Bolis M, Di Rito L, Pisati F, Giavazzi R, Zito E. Endoplasmic reticulum oxidoreductin 1-alpha deficiency and activation of protein translation synergistically impair breast tumour resilience. Br J Pharmacol. 2022;179:5180–95.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant numbers 82272489, 82203588); Mount Taishan Scholar Project Special Fund; Qingdao Traditional Chinese Medicine Science and Technology Project (2021-zyym28); and the Science and Technology Development Project of Shandong Geriatric Society (LKJGG2021W082). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
TYW and TL developed the concept, discussed the ideas, and wrote the manuscript. All authors made substantial, direct and intellectual contribution to the review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, T., Jiang, Y., Shi, W. et al. Endoplasmic reticulum stress: a novel targeted approach to repair bone defects by regulating osteogenesis and angiogenesis. J Transl Med 21, 480 (2023). https://doi.org/10.1186/s12967-023-04328-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04328-8