
Abstract
Background
Exosomes derived from stem cells have been widely studied for promoting regeneration and reconstruction of multiple tissues as “cell-free” therapies. However, the applications of exosomes have been hindered by limited sources and insufficient therapeutic potency.
Results
In this study, a stem cell-mediated gene therapy strategy is developed in which mediator mesenchymal stem cells are genetically engineered by bone morphogenetic protein-2 gene to produce exosomes (MSC-BMP2-Exo) with enhanced bone regeneration potency. This effect is attributed to the synergistic effect of the content derived from MSCs and the up-regulated BMP2 gene expression. The MSC-BMP2-Exo also present homing ability to the injured site. The toxic effect of genetical transfection vehicles is borne by mediator MSCs, while the produced exosomes exhibit excellent biocompatibility. In addition, by plasmid tracking, it is interesting to find a portion of plasmid DNA can be encapsulated by exosomes and delivered to recipient cells.
Conclusions
In this strategy, engineered MSCs function as cellular factories, which effectively produce exosomes with designed and enhanced therapeutic effects. The accelerating effect in bone healing and the good biocompatibility suggest the potential clinical application of this strategy.
Graphical Abstract

Similar content being viewed by others
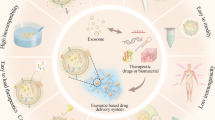

Introduction
The beneficial effects of mesenchymal stem cell-derived exosomes (MSC-Exo) in tissue regeneration have attracted significant interest in their applications in cell-free therapies [1]. Growing evidence has revealed that transplanted MSCs function rely on their paracrine actions through MSC-Exo in repairing injured tissue [2]. The positive roles of MSC-Exo have been proven in the repair and reconstruction of multiple tissues, including cartilage, skin, and skeleton [56]. Meanwhile, cells actively and selectively sort components in the cytoplasm to MVBs. Although the process of how specific cells identify these components remains unclear, our study demonstrated that MSCs could sort plasmid DNAs presented in the cytoplasm and expel them through exosome secretion. Interestingly, this mechanism functions as a re-encapsulation process, through which plasmid DNAs are equipped with a more biocompatible exosomal vehicle (Fig. 10). MVBs have two destinations within cells: merging with cell membranes to release exosomes or merging with lysosomes for degradation [57]. We found that by lysosomal inhibition, the yield of exosomes containing plasmid DNA could be significantly increased. This phenomenon might be applied in the mass production of plasmid DNA/exosome complexes, as substitutional vehicles for gene therapies.
This study developed a strategy of MSC-GT through producing exosomes with designed therapeutic effects. By selecting the proper combinations of transfection tools, gene sequences, and mediator stem cells, this strategy might find applications in the treatments of many diseases. It provides advantages of biocompatibility and bioavailability which may overcome the limitations confronting stem cell therapy, gene therapy and nanomedicine. Moreover, exosomes as nature nanocarriers with intriguing endogenous biofunctionalities and versatile biocomponents triggered the concept of establishing next-generation nanobiotechnology [58, 59]. However, the heterogeneity in living organisms and ambiguity of interacting mechanism with complicated in vivo milieu are the main obstacles impeding their clinical translations [60]. Future progress in isolation technologies and elucidating biological functionalities of exosome-based nanoplatforms would made it possible to envisage highly promising therapeutic applications.
Conclusions
In the present study, a stem cell-mediated gene therapy strategy is developed to produce exosomes (MSC-BMP2-Exo) with enhanced bone regeneration potency. Mesenchymal stem cells were genetically engineered by bone morphogenetic protein-2 gene to alter the content of the secreted exosomes. The enhanced bone regeneration effect is attributed to the synergistic effect of the content derived from MSCs and the up-regulated BMP2 expression. The MSC-BMP2-Exos also present biocompatibility, homing ability to the injured site, and plasmid DNA delivering. The accelerating effect in bone healing and the good biocompatibility suggest the potential clinical application of this strategy.
Material and methods
Animals
Male C57BL/6 mice (8 weeks old, 25 g) were purchased from Guangdong Medical Laboratory Animal Center (Guangdong, China). BALB/c nude mice (8 weeks old, 20 g) were obtained from Bei**g Vital River Laboratory (Bei**g, China). Mice were housed with access to water and food.
Cell culture
Human fetal bone marrow derived mesenchymal stem cells (hMSCs) were purchased from Cyagen Biosciences (HUXMA-01001, USA). Cells between passages three and ten were used and propagated in α-MEM (Gibco, USA) supplemented with 20% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin–streptomycin (Gibco, USA). Mouse hepatocytes were isolated from 8 weeks old male C57 mice, and cultured in L-DMEM (Hyclone, USA) containing 10% FBS and 2% penicillin–streptomycin [61]. Cells were maintained in a humidified atmosphere containing 95% air and 5% CO2 at 37 °C.
Cell transfection
Plasmid pIRES2-ZsGreen1-hBMP2 (pGFP-BMP2) encoding both BMP2 and GFP was constructed as previously described [62]. Plasmid pCI-neo-BMP2 (pBMP2) encoding BMP2 gene maintained in the lab was used for hMSCs genetically engineering. The hMSCs were transfected with plasmid using Lipofectamine 3000 transfection kit (Invitrogen, USA) according to the manufacturer’s instructions. Briefly, the hMSCs were seeded at a density to ensure 70–90% confluence after a 2-day culture. Equal amounts of Lipofectamin 3000 and P3000 reagent were diluted (1:50 v/v) independently and the plasmid was added into diluted P3000 (1:100 w/v). After incubation, above mentioned reagents were mixed and added to cells. The GFP expression in hMSCs was observed on a fluorescence microscope and quantified by flow cytometry (FACS Aria III, BD Biosciences, USA) 48 h post transfection. For intracellular tracking of plasmid in hMSCs during transfection, the plasmid was labeled with POPO-3 iodide (Invitrogen, USA) and observed under a fluorescence microscope at 2, 6, 24, and 48 h after transfection. For the lysosomal inhibitor assay, the medium was refreshed with α-MEM containing 10% exosome-free FBS (VivaCell, China) and 25 μM chloroquine diphosphate salt (Sigma, USA) 6 h after transfection. The exosomes were extracted at 24 and 48 h after cell transfection and incubated with aldehyde/sulfate latex beads (4 μm in diameter) before performing flow cytometry.
BMP2 concentration analysis by ELISA assay
After transfection, hMSC supernatants were collected for 48 h and centrifuged at 2000 × g for 20 min to remove cell debris. BMP2 protein levels of transfected and untransfected hMSCs were evaluated by Human BMP2 ELISA kit (Animalunion, China) according to the instructions.
Exosome isolation
Cells were washed three times with sterile phosphate buffer saline (PBS, Hyclone, USA), and cultured in α-MEM containing 10% exosome-free FBS. After 48 h, the supernatants were harvested and centrifuged at 2000 × g for 30 min to remove cells and debris. The total exosome isolation reagent (TEI, Thermo Scientific, USA) was used to purify exosomes according to the manufacturer's recommendations. To obtain a homogeneous solution, the mixture of above supernatant and reagent (2:1 v/v) was pipetted up and down 30 times and incubated overnight at 4 °C. The mixture was further centrifuged for 1 h at 10 000 × g at 4 °C. The supernatant was then aspirated and discarded, and the exosomes were resuspended with sterile PBS and stored at − 80 °C for further analysis [63].
Transmission electron microscopy and particle analysis
The exosomes were dropped separately on copper grids, and stained with 2% phosphotungstic acid (pH 7.0, Leagene Biotechnology, China) for 30 s. The staining solution were subsequently removed, and the copper grids loaded with exosomes were air dried overnight. Exosomes were detected using a transmission electron microscope (FEI, Tecnai G2 F20 S-TWIN, USA). For particle size and distribution, exosome suspension was diluted and detected by NTA using Zeta View (Particle Metrix, Germany).
Zeta potential analysis
MSC-BMP2-Exo or MSC-Exo at 0 day (freshly isolated) and 3 day (stored at − 80 °C) were suspended in PBS at the concentration of 0.5 mg/ml and zeta potential were measured by Zetasizer (NANO ZS, Malvern, UK).
Western blot analysis
Purified exosome pellets were lysed with RIPA lysis buffer (Beyotime, China) and PMSF (Beyotime, China). The cells were harvested and lysed using whole cell lysis kit (Keygen biotech, China). The protein concentrations were determined using a BCA protein assay kit (Thermo Scientific, USA). After boiling, equal amounts of proteins (20 μg) from different samples were separated by 10% SDS polyacrylamide gels and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, USA). The membrane was then blocked with 5% skim milk for 60 min at room temperature and incubated with different primary antibodies overnight at 4 °C, including anti-CD63 (Abcam, USA), anti-CD9 (Abcam, USA), anti-TSG 101 (Abcam, USA), anti-osterix (Abcam, USA), and anti-Runx2 antibodies (Abcam, USA). The PVDF membrane was further incubated with horseradish peroxidase-tagged secondary antibodies separately for 1 h. Protein bands were visualized using enhanced chemiluminescence assay (ECL, Amersham Biosciences, USA) and imaged by the Bio-RAD ChemiDoc gel imaging system (ChemiDoc XRS + system, Bio-Rad, USA).
Exosome labeling and cellular uptake
3,3’-dioctadecyloxacarbocyanine perchlorate (Dio)was used to label the exosomes (Beyotime, China). Purified exosomes were incubated with Dio (5 mM) for 15 min at 37 °C in the dark and then ultracentrifuged at 120 000 × g for 90 min to remove the unbounded dyes, followed by washing twice with PBS. The labeled exosome pellets were resuspended in PBS prior to use and hMSCs were cultured in a concentration of 1 × 105 cell/mL medium and incubated with Dio-labeled exosomes (80 μg/mL) overnight. DAPI was used to stain the cell nucleus. Intracellular localization of Dio-labeled exosomes was analyzed using a confocal laser scanning microscope (LSM 880, ZEISS, Germany), and quantified by flow cytometry.
CCK8 assay
Human fetal bone marrow derived mesenchymal stem cells (hMSCs) and hepatocytes were seeded in 96-well plates at a density of 1 × 104 cells per well and allowed to attach overnight. MSC-BMP2-Exo or MSC-Exo (2 μg per well, 100 μl) was incubated with cells for 72 h, and this concentration was adopted according to the reference [34]. For liposome group, the cells were transfected with plasmid-lipid complex (0.1 μg plasmid per well) according to the instructions of Lipofectamine 3000 kit for 6 h and refreshed with culture medium. The cellular viability was detected using a CCK-8 kit assay (Do**do, Japan) according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate reader (Thermo Scientific, USA). Experiments were repeated at least three times.
Osteogenic induction of hMSCs
Cells were planted in a 12-well cell culture dish (Corning, USA) at approximately 4 × 105 cell per well. After reaching 70–80% confluence, osteogenic differentiation was induced with the addition of osteogenic medium, consisting of α-MEM medium supplemented with 10% FBS, 1% penicillin streptomycin solution, 50 μg/mL L-ascorbic acid-2-phosphate (Sigma, USA), 10 mM β-glycerophosphate (Sigma, USA), and 10 nM dexamethasone (Sigma, USA). Exosomes were added with 30 μg per well (20 μg/ml) [64] and the osteogenic medium was changed every three days.
Quantitative real-time PCR
The total RNAs from different kinds of cells were extracted using TRIzol (Invitrogen, USA). The purity and concentration of RNAs were detected by measuring the absorbance on a NanoDrop2000 (Thermo Scientific, USA) at 260 and 280 nm. The samples with ratios from 1.8 to 2.0 were then qualified for the next reverse transcription reaction. An IScript cDNA Synthesis Kit (Thermo Scientific, USA) was used to synthesize cDNAs according to the manufacturer’s instructions. Each real-time PCR was prepared in a 20 μL of reaction mixture and performed on a CFX96 Real-Time system (Bio-rad, USA). Loading control was β-actin. PCR primer sequences for gene expression analyses are listed in Table 1. PCR products were proved by melting curve analysis.
Alkaline phosphatase and Alizarin red staining
Cells were induced for osteogenic differentiation and supplemented with different exosomes. Alkaline phosphatase was stained using a BCIP/NBT alkaline phosphatase color development kit (Beyotime, China) 2 weeks after culture. The deposition of calcium phosphate was stained with Alizarin Red S (1%, pH 4.2, Solarbio, China) 3 weeks after differentiation. The images were taken by a biological microscope (Olympus, BX53, Japan). Mineralized matrix was quantified by dissolving Alizarin Red S in cetylpyridinium chloride solution (100 mM, Sigma, USA) for 1 h, and read on a multi-plate reader (Multiskan FC Microplate Photometer, Thermo Fisher Scientific, USA) at 562 nm.
FACS analysis of exosomes
Exosomes were coated onto beads (MW3000, Invitrogen, USA) and FACS analysis was performed. Exosomes (20 μg) were incubated with aldehyde/sulfate latex beads (5 μL, 4 μm in diameter) for 15 min at room temperature. The bead/exosome mixture was then diluted with 1 mL of PBS and incubated for another 2 h at room temperature under gentle shaking. The beads were then spun down for 3 min at 4000 × g, washed with PBS, and resuspended in FACS buffer. The beads were analyzed by flow cytometry using a FACS-Calibur flow cytometer and FlowJo software.
Mouse femoral defect model and exosome treatment
Bone defects were created at femora to evaluate the bone regeneration effects of exosomes. C57BL/6 male mice were anesthetized with an intraperitoneal injection of 10% chloral hydrate and kept on a warming pad throughout the surgical procedure. The right hindlimb was shaved and aseptically prepared for surgery by disinfecting with 75% ethanol. Skin incisions of approximately 5 mm were made on the right hind limbs from the lateral side, and femora were exposed by splitting the muscle. For the trabecular bone defect model, 1 mm-diameter holes were created at the lateral femoral distal metaphysis using an orthopedic electric drill. The holes were rinsed by injection of saline using a 1 ml syringe to discard bone fragments from the cavity. The incised muscle and skin were closed with nylon sutures. Perforations did not cause significant perioperation or post-operation fractures. Thirty-six mice with femoral trabecular bone defects were divided into three groups. Each group was treated with either 0.9% saline, MSC-Exo, or MSC-BMP2-Exo. Exosomes (50 μg, 1.8 μg/uL) were injected in situ to the bone defect for the first time on the third day after surgery [65], and treated once a week. The same volume of saline was injected as a control group. At each time point, 15 and 30 days after the first exosome treatment, mice were killed by cervical dislocation. Excised femora were fixed in 10% neutral buffered formalin for 24 h at 4 °C in the dark. After 24 h, the samples were rinsed with running water for about 1 h, then transferred into PBS and stored at 4 °C for further micro-CT and immunohistochemical analysis of trabecular bone defect model. For the cortical bone defect model, holes of the same diameter were created at the mid-diaphysis of the femora. Fifteen mice were divided into three groups, and treated with either 0.9% saline, MSC-Exo, or MSC-BMP2-Exo, with five mice in each group. Exosomes were administrated similarly. In vivo micro-CT imaging of cortical bone defect model was performed at 15 and 30 days after the first exosome treatment.
Micro-CT detection
Mouse femora were separated, and the soft tissue was removed. Bone quality was analyzed using high-resolution micro-CT (SkyScan1176, Belgium). Scanning was performed at a voltage of 60 kV, a current of 417 µA, and a resolution of 9.0 µm/pixel. Software of NRecon, CTAn and μCTVol were used for three-dimensional reconstruction (threshold 71) and parameter analysis. For the cancellous bone defect, 400 slices in the region of distal femur above the growth plate were selected for three-dimensional reconstruction, and a cubical region with 70 slices in the center of defect was chosen for structural parameter analysis. The structural parameter of the trabecular bone, including bone mineral density (BMD), bone volume fraction (BV/TV), trabecular number (Tb.N), and trabecular thickness (Tb.Th), were calculated through model-independent 3D measurement. For the cortical bone defect in the femur, 340 continuous slices and 20 slices with the middle slice in the center of defect were chosen for three-dimensional reconstruction. The isolated cortical region in the defect region with 110 slices was analyzed for BV/TV and cortical thickness (Cr.Th).
Histological and immunohistochemical analysis
After the micro-CT imaging and analysis were performed, the excised femora were decalcified using 10% EDTA (pH = 7.4) at room temperature for 2 weeks. The samples were dehydrated using a gradient ethanol series and a final xylene step and were subsequently paraffin embedded. Approximately 5-μm-thick sections were made. Sectioning of the paraffin-embedded samples was performed along the longitudinal axis of the femur. Longitudinal sections were prepared using a microtome (Leica, USA) and tungsten carbide blades. Sections were stained with hematoxylin–eosin and saffran (Beyotime, China). Images of the femur defect region were acquired via light microscopy. For immunohistochemical analysis, bone sections were incubated with primary antibodies against anti-phospho Smad1/5/8 (Millipore, USA) overnight at 4 °C, and then covered with secondary antibodies (Servicebio, China) at room temperature for 50 min. After the sections were cleaned in PBS, DAB color develo** solution (Servicebio, China) was used for color development. The slices were flushed with tap water to terminate color development. The slices were stained with hematoxylin for approximately 3 min and rinsed with water. Finally, dehydrated seal was performed, and the images were collected and analyzed with a microscope.
Biodistribution of exoxomes in mice
Exosomes were labeled with Cy5.5 for in vivo fluorescence imaging. Exosomes were incubated with Cy5.5 NHS ester dye (10 mM, Abcam, USA) for 2 h in the dark. The unbound dye was then removed by three washing steps on 100 kDa ultrafiltration tubes (Millipore, USA). The mouse bone defect model was established, as previously mentioned above, on the right distal femur. Exosomes labeled with Cy5.5 (50 μg each leg) were injected in situ at the distal femur on both sides with and without bone defect on the second day after the establishment of the bone defect model. Six nude mice were divided into two groups for MSC-Exo and BMP2-MSC-Exo. In vivo fluorescence images were obtained using an IVIS imaging system (IVIS Spectrum, PerkinElmer, USA). The distribution of Cy5.5-labeled exosomes was observed at 6, 24, or 48 h after injection. The ex vivo fluorescence images were then visualized from the sacrificed mice using the IVIS imaging system. The fluorescence signals were normalized to track the change tendency within a group. To investigate the biodistribution of exosomes in different organs, exosomes labeled with Cy5.5 (100 μg each mouse) were intravenously injected through the tail vein of mice with bone defects on the right distal femur. Mice without bone defects were used for comparison. These mice were sacrificed 48 h after injection, with 3 mice in each group. The heart, lungs, liver, spleen, kidneys and legs were harvested for IVIS imaging.
Statistical analysis
All experiments were performed in at least three independent cultures/animals per genotype, treatment, and condition. Statistics were assessed using the GraphPad Prism 8 Software and results are presented as mean ± standard deviations (SD). Comparison between two groups was conducted by t-test, whereas multigroup comparisons were conducted by one-way analysis of variance with Tukey’s post hoc test. P-values less than 0.05 were considered significant. (*p < 0.05; **p < 0.01; ***p < 0.001).
Availability of data and materials
The data and materials of the study are available from the corresponding author on reasonable request.
References
Varderidou-Minasian S, Lorenowicz MJ. Mesenchymal stromal/stem cell-derived extracellular vesicles in tissue repair: challenges and opportunities. Theranostics. 2020;10:5979–97.
Jiang S, Tian G, Yang Z, Gao X, Wang F, Li J, Tian Z, Huang B, Wei F, Sang X, Shao L, Zhou J, Wang Z, Liu S, Sui X, Guo Q, Guo W, Li X. Enhancement of acellular cartilage matrix scaffold by Wharton’s jelly mesenchymal stem cell-derived exosomes to promote osteochondral regeneration. Bioact Mater. 2021;6:2711–28.
Xu X, Liang Y, Li X, Ouyang K, Wang M, Cao T, Li W, Liu J, **ong J, Li B, **a J, Wang D, Duan L. Exosome-mediated delivery of kartogenin for chondrogenesis of synovial fluid-derived mesenchymal stem cells and cartilage regeneration. Biomaterials. 2021;269:120539.
Duan MN, Zhang Y, Zhang HY, Meng YP, Qian M, Zhang GK. Epidermal stem cell-derived exosomes promote skin regeneration by downregulating transforming growth factor-beta 1 in wound healing. Stem Cell Res Ther. 2020;11:452.
Liu A, Lin D, Zhao H, Chen L, Cai B, Lin K, Shen SGF. Optimized BMSC-derived osteoinductive exosomes immobilized in hierarchical scaffold via lyophilization for bone repair through Bmpr2/Acvr2b competitive receptor-activated Smad pathway. Biomaterials. 2021;272:120718.
Hu SQ, Li ZH, Shen DL, Zhu DS, Huang K, Su T, Dinh PU, Cores J, Cheng K. Exosome-eluting stents for vascular healing after ischaemic injury. Nat Biomed Eng. 2021;5:1174–88.
Mendt M, Rezvani K, Shpall E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transplant. 2019;54:789–92.
Zhai MM, Zhu Y, Yang MY, Mao CB. Human Mesenchymal stem cell derived exosomes enhance cell-free bone regeneration by altering their miRNAs profiles. Adv Sci. 2020;7:2001334.
Brennan MA, Layrolle P, Mooney DJ. Biomaterials functionalized with MSC secreted extracellular vesicles and soluble factors for tissue regeneration. Adv Funct Mater. 2020;30:1909125.
Malda J, Boere J, van de Lest CHA, van Weeren PR, Wauben AHM. Extracellular vesicles—new tool for joint repair and regeneration. Nat Rev Rheumatology. 2016;12:243–9.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:640.
Zhou B, Xu K, Zheng X, Chen T, Wang J, Song Y, Shao Y, Zheng S. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct Target Ther. 2020;5:144.
Lei Q, Gao F, Liu T, Ren WX, Chen L, Cao YL, Chen WL, Guo SJ, Zhang Q, Chen WQ, Wang HX, Chen ZC, Li QB, Hu Y, Guo AY. Extracellular vesicles deposit PCNA to rejuvenate aged bone marrow-derived mesenchymal stem cells and slow age-related degeneration. Sci Transl Med. 2021;13:eaaz8697.
Bjorge IM, Kim SY, Mano JF, Kalionis B, Chrzanowski W. Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine—a new paradigm for tissue repair. Biomater Sci. 2017;6:60–78.
Mittelbrunn M, Sanchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. 2012;13:328–35.
Mercier V, Larios J, Molinard G, Goujon A, Matile S, Gruenberg J, Roux A. Endosomal membrane tension regulates ESCRT-III-dependent intra-lumenal vesicle formation. Nat Cell Biol. 2020;22:947–59.
McNally EK, Brett CL. The intralumenal fragment pathway mediates ESCRT-independent surface transporter down-regulation. Nat Commun. 2018;9:5358.
Zhao LW, Gu CY, Gan Y, Shao LL, Chen HW, Zhu HY. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J Control Release. 2020;318:1–15.
Yim N, Ryu SW, Choi K, Lee KR, Lee S, Choi H, Kim J, Shaker MR, Sun W, Park JH, Kim D, Heo WD, Choi C. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun. 2016;7:12277.
Richter M, Vader P, Fuhrmann G. Approaches to surface engineering of extracellular vesicles. Adv Drug Deliv Rev. 2021;173:416–26.
Du W, Zhang KY, Zhang SQ, Wang R, Nie Y, Tao HY, Han ZB, Liang L, Wang D, Liu JF, Liu N, Han ZC, Kong DL, Zhao Q, Li ZJ. Enhanced proangiogenic potential of mesenchymal stem cell-derived exosomes stimulated by a nitric oxide releasing polymer. Biomaterials. 2017;133:70–81.
Huang CC, Kang MY, Lu Y, Shirazi S, Diaz JI, Cooper LF, Gajendrareddy P, Ravindran S. Functionally engineered extracellular vesicles improve bone regeneration. Acta Biomater. 2020;109:182–94.
Venkatesan JK, Rey-Rico A, Meng W, Cai X, Pons F, Lebeau L, Migonney V, Madry H, Cucchiarini M. Biomaterial-assisted gene therapy for translational approaches to treat musculoskeletal disorders. Mater Today Adv. 2021;9:100126.
Yang Z, Shi J, **e J, Wang Y, Sun J, Liu T, Zhao Y, Zhao X, Wang X, Ma Y, Malkoc V, Chiang C, Deng W, Chen Y, Fu Y, Kwak KJ, Fan Y, Kang C, Yin C, Rhee J, Bertani P, Otero J, Lu W, Yun K, Lee AS, Jiang W, Teng L, Kim BYS, Lee LJ. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat Biomed Eng. 2020;4:69–83.
Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, ** J, Liu Q, Evans R, Fissell WH, Patton JG, Rome LH, Burnette DT, Coffey RJ. Reassessment of exosome composition. Cell. 2019;177:428–45.
Witwer KW, Wolfram J. Extracellular vesicles versus synthetic nanoparticles for drug delivery. Nat Rev Mater. 2021;6:103–6.
Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014;3:23743.
Ullah M, Kodam SP, Mu Q, Akbar A. Microbubbles versus extracellular vesicles as therapeutic cargo for targeting drug delivery. ACS Nano. 2021;15:3612–20.
Luan X, Sansanaphongpricha K, Myers I, Chen H, Yuan H, Sun D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol Sin. 2017;38:754–63.
Liao W, Du Y, Zhang CH, Pan FW, Yao Y, Zhang T, Peng Q. Exosomes: the next generation of endogenous nanomaterials for advanced drug delivery and therapy. Acta Biomater. 2019;86:1–14.
Zhu XH, Badawi M, Pomeroy S, Sutaria DS, **e ZL, Baek A, Jiang JM, Elgamal OA, Mo XK, La Perle K, Chalmers J, Schmittgen TD, Phelps MA. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6:1324730.
Sun L, Fan M, Huang D, Li B, Xu R, Gao F, Chen Y. Clodronate-loaded liposomal and fibroblast-derived exosomal hybrid system for enhanced drug delivery to pulmonary fibrosis. Biomaterials. 2021;271:120761.
Han Z, Liu SQ, Pei YG, Ding Z, Li YG, Wang XG, Zhan DQ, **a SL, Driedonks T, Witwer KW, Weiss RG, van Zijl PCM, Bulte JWM, Cheng LZ, Liu GS. Highly efficient magnetic labelling allows MRI tracking of the homing of stem cell-derived extracellular vesicles following systemic delivery. J Extracell Vesicles. 2021;10:e12054.
Zhan Q, Yi K, Li X, Cui X, Yang E, Chen N, Yuan X, Zhao J, Hou X, Kang C. Phosphatidylcholine-engineered exosomes for enhanced tumor cell uptake and intracellular antitumor drug delivery. Macromol Biosci. 2021;21:2100042.
Garcia-Arranz M, Herreros MD, Gonzalez-Gomez C, de la Quintana P, Guadalajara H, Georgiev-Hristov T, Trebol J, Garcia-Olmo D. Treatment of Crohn’s-related rectovaginal fistula with allogeneic expanded-adipose derived stem cells: a phase I-IIa clinical trial. Stem Cells Transl Med. 2016;5:1441–6.
Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194–200.
Heslop JA, Hammond TG, Santeramo I, Piella AT, Hopp I, Zhou J, Baty R, Graziano EI, Marco BP, Caron A, Skold P, Andrews PW, Baxter MA, Hay DC, Hamdam J, Sharpe ME, Patel S, Jones DR, Reinhardt J, Danen EHJ, Ben-David U, Stacey G, Bjorquist P, Piner J, Mills J, Rowe C, Pellegrini G, Sethu S, Antoine DJ, Cross MJ, Murray P, Williams DP, Kitteringham NR, Goldring CEP, Park BK. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cell Transl Med. 2015;4:389–400.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
**ng HY, Zhang ZJ, Mao QJ, Wang CG, Zhou YL, Zhou XP, Ying LW, Xu HB, Hu SJ, Zhang N. Injectable exosome-functionalized extracellular matrix hydrogel for metabolism balance and pyroptosis regulation in intervertebral disc degeneration. J Nanobiotechnol. 2021;19:1–15.
Xu LZ, Faruqu FN, Lim YM, Lim KY, Liam-Or R, Walters AA, Lavender P, Fear D, Wells CM, Wang JTW, Al-Jamal KT. Exosome-mediated RNAi of PAK4 prolongs survival of pancreatic cancer mouse model after loco-regional treatment. Biomaterials. 2021;264:120369.
Li SP, Lin ZX, Jiang XY, Yu XY. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharm Sin. 2018;39:542–51.
Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–28.
Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nature Genet. 2006;38:1424–9.
Ambrosi TH, Marecic O, McArdle A, Sinha R, Gulati GS, Tong X, Wang Y, Steininger HM, Hoover MY, Koepke LS, Murphy MP, Sokol J, Seo EY, Tevlin R, Lopez M, Brewer RE, Mascharak S, Lu L, Ajanaku O, Conley SD, Seita J, Morri M, Neff NF, Sahoo D, Yang F, Weissman IL, Longaker MT, Chan CKF. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021;597:256–62.
Bez M, Pelled G, Gazit D. BMP gene delivery for skeletal tissue regeneration. Bone. 2020;137:115449.
Hsieh MK, Wu CJ, Chen CC, Tsai TT, Niu CC, Wu SC, Lai PL. BMP-2 gene transfection of bone marrow stromal cells to induce osteoblastic differentiation in a rat calvarial defect model. Mater Sci Eng C-Mater Biol Appl. 2018;91:806–16.
Szostak N, Royo F, Rybarczyk A, Szachniuk M, Blazewicz J, del Sol A, Falcon-Perez JM. Sorting signal targeting mRNA into hepatic extracellular vesicles. RNA Biol. 2014;11:836–44.
Kamal AF, Siahaan OSH, Fiolin J. Various dosages of BMP-2 for management of massive bone defect in Sprague Dawley rat. Arch Bone Jt Surg. 2019;7:498–505.
Yin B, Ma Q, Song C, Zhao L, Yu F, Wang C, Shi Y, Ye L. Exosome-derived noncoding RNAs as a promising treatment of bone regeneration. Stem Cells Int. 2021;2021:1–8.
Oliveira AC, Ferraz MP, Monteiro FJ, Simoes S. Cationic liposome-DNA complexes as gene delivery vectors: development and behaviour towards bone-like cells. Acta Biomater. 2009;5:2142–51.
Valente JFA, Pereira P, Sousa A, Queiroz JA, Sousa F. Effect of plasmid DNA size on chitosan or polyethyleneimine polyplexes formulation. Polymers. 2021;13:793.
Baghaei M, Tekie FSM, Khoshayand MR, Varshochian R, Hajiramezanali M, Kachousangi MJ, Dinarvand R, Atyabi F. Optimization of chitosan-based polyelectrolyte nanoparticles for gene delivery, using design of experiment: in vitro and in vivo study. Mater Sci Eng C-Mater Biol Appl. 2021;118:111036.
Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, Martin-Cofreces NB, Jaso-Tamame AL, Iborra S, Jorge I, Gonzalez-Aseguinolaza G, Garaude J, Vicente-Manzanares M, Enriquez JA, Mittelbrunn M, Sanchez-Madrid F. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun. 2018;9:2658.
Koltover I, Salditt T, Radler JO, Safinya CR. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science. 1998;281:78–81.
Rehman ZU, Hoekstra D, Zuhorn IS. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano. 2013;7:3767–77.
Morille M, Passirani C, Vonarbourg A, Clavreul A, Benoit JP. Progress in develo** cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29:3477–96.
Villarroya-Beltri C, Baixauli F, Mittelbrunn M, Fernandez-Delgado I, Torralba D, Moreno-Gonzalo O, Baldanta S, Enrich C, Guerra S, Sanchez-Madrid F. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat Commun. 2016;7:13588.
Kwon SH, Al Faruque H, Kee H, Kim E, Park S. Exosome-based hybrid nanostructures for enhanced tumor targeting and hyperthermia therapy. Colloid Surf B-Biointerfaces. 2021;205:111915.
Yang BW, Chen Y, Shi JL. Exosome biochemistry and advanced nanotechnology for next-generation theranostic platforms. Adv Mater. 2019;31:1802896.
Sharma S, Masud MK, Kaneti YV, Rewatkar P, Koradia A, Hossain MSA, Yamauchi Y, Popat A, Salomon C. Extracellular vesicle nanoarchitectonics for novel drug delivery applications. Small. 2021;17:2102220.
Yang M, Liu Q, Huang TL, Tan WJ, Qu LB, Chen TK, Pan HB, Chen L, Liu JS, Wong CW, Lu WW, Guan M. Dysfunction of estrogen-related receptor alpha-dependent hepatic VLDL secretion contributes to sex disparity in NAFLD/NASH development. Theranostics. 2020;10:10874.
Yue JH, Wu J, Liu D, Zhao XL, Lu WW. BMP2 gene delivery to bone mesenchymal stem cell by chitosan-g-PEI nonviral vector. Nanoscale Res Lett. 2015;10:203.
Dash M, Palaniyandi K, Ramalingam S, Sahabudeen S, Raja NS. Exosomes isolated from two different cell lines using three different isolation techniques show variation in physical and molecular characteristics. Biochim Biophys Acta Biomembr. 2020;1863:183490.
Zha Y, Li Y, Lin T, Chen J, Zhang S, Wang J. Progenitor cell-derived exosomes endowed with VEGF plasmids enhance osteogenic induction and vascular remodeling in large segmental bone defects. Theranostics. 2021;11:397–409.
Gupta D, Zickler AM, El Andaloussi S. Dosing extracellular vesicles. Adv Drug Deliv Rev. 2021;178:113961.
Acknowledgements
Not applicable
Funding
This work was supported by the National Natural Science Foundation of China (81972071, 81802959); Guangdong Basic and Applied Basic Research Foundation (2021A1515010191, 2018A030313888); Science and Technology Research Funding of Shenzhen (JCYJ20210324102001003, JSGG20180507183242702, and JCYJ20200109150420892) and HKU-SZH Fund for Shenzhen Key Medical Discipline (SZXK2020084).
Author information
Authors and Affiliations
Contributions
FL, JW, DL and LH performed the experiments, analyzed data and wrote manuscript. YL performed part of in vitro experiments. DY, KY, DC, WL and HP provided technical assistance throughout the project. XZ conceived hypothesis, designed experiments, proposes and designs plasmids, revised the manuscript. XZ and TW coordinated projects and provided funding. All authors approved the final submitted version of this paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were carried out in accordance with the protocols approved by the Animal Ethics Committee of Institute of Advanced Technology, Chinese Academy of Sciences. (IACUC Number: SIAT-IACUC-20190307-YYS-TXXZX-ZXL-01).
Consent for publication
All authors agreed to publish this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. BMP2 concentration of hMSCs after transfected for 48 h (n=3), *p < 0.05. Figure S2. Changes of zeta potential of exosomes at 0 day and 3 day (n=3). Figure S3. Cell viability of hepatocytes incubated with exosomes was determined by CCK-8 assay (n=3).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, F., Wu, J., Li, D. et al. Engineering stem cells to produce exosomes with enhanced bone regeneration effects: an alternative strategy for gene therapy. J Nanobiotechnol 20, 135 (2022). https://doi.org/10.1186/s12951-022-01347-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-022-01347-3