
Abstract
Protein kinase C (PKC) and Protein kinase D (PKD) isoforms can sense diacylglycerol (DAG) generated in the different cellular compartments in various physiological processes. DAG accumulates in multiple organs of the obese subjects, which leads to the disruption of metabolic homeostasis and the development of diabetes as well as associated diseases. Multiple studies proved that aberrant activation of PKCs and PKDs contributes to the development of metabolic diseases. DAG-sensing PKC and PKD isoforms play a crucial role in the regulation of metabolic homeostasis and therefore might serve as targets for the treatment of metabolic disorders such as obesity and diabetes.
Similar content being viewed by others


Diacylglycerol (DAG) – the structure and sources
Diacylglycerol (DAG) is a neutral lipid involved in various metabolic pathways in the cell. It is an important component of membranes that also acts as a secondary messenger. It was shown that DAG is involved among others in multiple processes and pathways, e.g. protein transport, vesicle secretion, insulin signaling, cell growth, and proliferation [1,2,3]. In the cell, DAG can be either synthesized during de novo lipid biosynthesis or generated from other intracellular lipid species. Different reactions contributing to the generation of DAG occur in various subcellular compartments like plasma membrane, Golgi network, endoplasmic reticulum (ER), and lipid droplets. During de novo biosynthesis of triacylglycerol (TAG) and phospholipid (PL), DAG is produced as an intermediate by acyltransferases and phosphohydrolases [4, 5]. DAG is also generated from TAG stored in either cytoplasmic and ER-associated lipid droplets in a reaction catalyzed by lipases or in the plasma membrane and Golgi complex from PL by acyltransferases [6].

Sources of diacylglycerols (DAG) in the cell. Intracellularly, DAG is produced as an intermediate during de novo biosynthesis of triacylglycerol (TAG) and phospholipid (PL), and during catabolism of TAG stored in either cytoplasmic and endoplasmic reticulum-associated lipid droplets or PL in the plasma membrane and Golgi complex. Generated DAG locally recruits and promotes activation of conventional and novel protein kinases C (PKCs) and protein kinases D (PKDs). For detailed description see the text. ATGL – adipose triglyceride lipase, DAGK – diacylglycerol kinase, DAGL – diacylglycerol lipase, DGAT – diglyceride acyltransferase, ER – endoplasmic reticulum, HSL – hormone-sensitive lipase, MAG – monoacylglycerol, MGAT – monoacylglycerol-O-acyltransferase, PA – phosphatidic acid, PC – phosphatidylcholine, PIP2 – phosphatidylinositol 4,5-bisphosphate, PLC – phospholipase, SMS – sphingomyelin synthase

The role of PKC and PKD isoforms in various tissues and organs
DAG consists of two fatty acids chains covalently bound to glycerol through ester bonds. There are three different isomeric forms of DAG: sn-1,2, sn-2,3 and rac-1,3 DAG [7]. It was shown that different enzymes could discriminate between DAG isomers [3, 8]. Moreover, proteins interacting with DAG, depending on their function, are localized in different subcellular compartments. Thus, it was suggested that DAG stereo/regioisomers are localized in distinct parts of the cell and play different roles in cell signaling [6, 7]. DAG can be generated from TAG during the first step of lipolysis. During this process, TAG is hydrolyzed into fatty acid and DAG by TAG lipases [6]. TAG stored in cytoplasmic and ER-associated lipid droplets is accessible to different kinds of lipases. Adipose triglyceride lipase (ATGL) is the primary enzyme responsible for the hydrolysis of TAG localized in the cytoplasm. It generates rac-1,3 DAG and is the only known lipase that can hydrolyze TAG at the sn-2 position. Moreover, in the presence of its activator - comparative gene identification-58, ATGL can also hydrolyze TAG to sn-2,3 DAG [3]. Another enzyme that can hydrolyze cytoplasmic TAG is hormone-sensitive lipase (HSL), which produces sn-2,3 DAG. At the same time, DAG generated on cytoplasmic lipid droplets is a substrate for HSL and diacylglycerol lipase β (DAGLβ), which hydrolase it to fatty acid and monoacylglycerol (MAG). It was shown that HSL specifically hydrolyzes rac-1,3 DAG and sn-2,3 DAG and shows a preference for polyunsaturated fatty acids [8, 9]. Cytoplasmic DAG can be also re-esterified to TAG via diglyceride acyltransferase (DGAT2). At ER and Golgi network, DAG is generated as mentioned above during TAG hydrolysis, but also through de novo biosynthesis from MAG or phosphatidic acid (PA) in reactions catalyzed by monoacylglycerol-O-acyltransferase (MGAT) and PA phosphohydrolase respectively or as a side product during sphingomyelin synthesis. The main isomeric form present at ER and Golgi network is sn-1,2 DAG, which can be metabolized by DGAT1 and DGAT2 to TAG, diacylglycerol kinase (DAGK) to PA or enzymes involved in PL formation [3, 10, 11]. At the plasma membrane, sn-1,2 DAG is generated during sphingomyelin synthesis or cleavage of phosphoglycerol from glycerophospholipids in a reaction catalyzed by phospholipase C (PLC) [3, 12]. Phosphatidylinositol-specific PLC (PI-PLC) hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) and generates 1,2-sn DAG, which specifically activates protein kinase C and subsequently protein kinase D [2, 3, 13].
DAG is not only generated intracellularly but is also an intermediate of extracellular lipid metabolism and can be supplied with diet. Extracellular DAG is a product of TAG hydrolysis during digestion and in the catabolism of lipoprotein-associated TAG in the bloodstream. Since DAG generated in the digestive system or in circulation is usually immediately hydrolyzed to MAG and fatty acid, it is probably not involved in the regulation of signaling pathways.
Intracellular changes in DAG level are affecting various signaling pathways and processes. For instance, an elevated level of DAG in many tissues is correlated with impaired cell metabolism and pathogenesis of metabolic disorders like insulin resistance. Additionally, a high-fat diet (HFD) feeding in animal models leads to the development of metabolic disorders accompanied by an increased level of DAG in peripheral tissues [14]. ATGL-deficiency in mice that leads to decreased DAG level and increased TAG content, protects from HFD-induced insulin resistance and glucose intolerance [15]. This finding supports the statement that DAG can disrupt insulin signaling in peripheral tissues. The described phenomenon is caused by DAG acting as a secondary messenger through interactions with several proteins in the cell. It was shown that DAG can interact with proteins containing the C1 domain that represents the recognition motif for DAG and phorbol esters. C1 domain was found among others in protein kinases C (PKCs), protein kinases D (PKDs), DAGKs, Rac GTPase-activating proteins (Rac-GAPs), Ras guanyl nucleotide-releasing proteins (RasGRPs), and mammalian uncoordinated-13 proteins (Munc13s) [16,17,18,19,20]. Different proteins within the families containing the C1 domain differ in affinity to DAG and phorbol esters. However, DAG is involved in the regulation of various signaling pathways through interaction with these proteins. Due to a large amount of information available up to date about different classes of DAG-sensing proteins involved in regulation of various physiological processes, this review is focused only on DAG-dependent PKCs and PKDs kinases which DAG directly activates, and their function in the regulation of lipid and glucose metabolism in various tissues.
PKC isoforms – structure and classification
PKCs are a family of serine/threonine kinases involved in various processes in cells including proliferation, differentiation, cell survival, and apoptosis [21, 22]. Moreover, PKC plays a vital role in the pathogenesis of such diseases like diabetes or cancer [23, 24]. PKC family is composed of three different subgroups: conventional (cPKC), the novel (nPKC) and atypical (aPKC). PKCα, β1, β2, and γ belong to cPKC, PKCδ, ε, η and θ are nPKC, whereas aPKC comprises of PKCζ and λ/ι. All PKCs consist of the N-terminal regulatory region and C-terminal catalytic region (kinase domain) [1]. The regulatory region of all PKCs contains an autoinhibitory pseudosubstrate segment. Binding of secondary messengers or proteins scaffolds to specific modules in the regulatory region controls the position of the pseudosubstrate segment in or out of the substrate-binding site. Therefore it regulates the activity of the PKCs [1]. All of the PKCs contain at least one C1 domain with different affinity for DAG. Both cPKC and nPKC contain two C1 domains: C1A and C1B. However, it was shown that the C1B in nPKC has a much higher affinity to DAG than C1B in cPKC [25]. In both subgroups, C1B is considered as a primary DAG sensor [26]. Thus, nPKC respond to an increase in DAG content alone, whereas cPKC needs additional increase in intracellular Ca2+ to be fully activated [25]. The C1 domain of aPKC has no affinity to DAG and acts as a part of the autoinhibitory segment [27]. cPKC and nPKC are activated specifically via 1,2-sn DAG, usually generated by PLC [7]. cPKC also contains Ca2+-sensing C2 domain, which binds PIP2 in the plasma membrane. Similarly, C2 domain is present in nPKC. However, this domain cannot sense Ca2+ and bind PIP2 [1]. aPKC contains the PB1 domain, which mediates binding to protein scaffolds [27].
PKCs are physiologically activated by various extracellular signals transduced by hormones, growth factors, cytokines or antigens [28]. cPKC and nPKC respond to the elevation of intracellular DAG, which binds to PKCs and causes their activation and translocation to the membranes. Ca2+-dependent signals lead to rapid activation of cPKC, whereas nPKC may also be activated at the Golgi network and other cellular membranes in a more sustainable manner [29]. The presence of activated PKCs on internal membranes leads to the phosphorylation of various interacting proteins. For instance in vitro studies and experiments performed in cells expressing insulin receptor showed that PKCs phosphorylate specific serine/threonine residues of insulin receptor and its intracellular substrates belonging to the family of insulin receptor substrates (IRSs), causing impairment of insulin signaling pathway [30,31,32,33,34,35]. Abundance and role in metabolism regulation of different members of the PKC family differ depending on the type of tissue and will be further discussed below.
PKDs – downstream effectors of PKCs and DAG
Similarly to PKCs, PKDs are DAG-activated family of serine/threonine kinases involved in various processes and pathways in the cell. However, substrate specificity varies between PKCs and PKDs and they play different roles in cell signaling. PKDs are calmodulin-dependent kinases and they regulate such processes as vesicle trafficking, cell differentiation, motility and apoptosis [36,37,38]. PKD family is composed of three isoforms: PKD1, PKD2, and PKD3. All PKDs consist of N-terminal regulatory domains (C1 domains and autoinhibitory PH domain) and the C-terminal catalytic region (kinase domain). PKDs contain two DAG-binding C1 domains: C1a and C1b, but DAG preferably binds to the C1a domain [39]. At N-terminus PKD1 and PKD2, but not PKD3 contain additionally hydrophobic Ala(/Pro)-rich region, which potentially can be inserted into the membranes [40]. Moreover, at C-terminus PKD1 and PKD2 contain a PDZ-binding motif with an autophosphorylation site [41].
PKDs are activated by extracellular signals transduced by hormones, growth factors, cytokines, and neurotransmitters [38]. DAG in cell membranes recruits PKD through its C1 domains and induces conformational changes that abrogate PKDs autoinhibitory mechanism, leads to autophosphorylation at C-terminus of PKD1 and PKD2, and subsequently activates PKDs [2, 42]. Next, DAG-activated PKC phosphorylates serine residues in the PKDs activation loop located in the kinase domain, which in turn causes further conformational changes resulting in the maximal increase of catalytic activity of PKDs [38]. PKC phosphorylates Ser738 and Ser742, Ser706 and Ser710, Ser731 and Ser735 in human PKD1, PKD2 and PKD3, respectively [2]. Both cPKC and nPKC can activate PKDs, however, it was shown that PKDs are phosphorylated mainly by nPKC [43, 44]. Prolonged presence of extracellular stimuli leads to autophosphorylation of a serine residue in the PKDs activation loop, therefore to PKC-independent activation of PKDs and induction of long-term effects of PKD on cell signaling [45, 46]. PKDs can be found in different cell compartments, including Golgi and plasma membranes, nucleus, cytoplasm, and mitochondria. Their activity depends on the availability of local DAG. PKDs are widely expressed in mammals but their level and specific role in cell signaling can vary between tissues, which will be further discussed below.
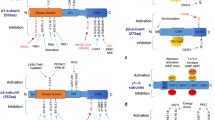
DAG-evoked activation of PKCs and PKDs suppresses insulin signaling in hepatocytes
Liver plays a central role in metabolic homeostasis by regulating glucose, lipid and protein metabolism. Under physiological conditions such as feeding, hepatic insulin stimulation promotes glucose uptake, lipogenesis and inhibits gluconeogenesis. Conversely, upon fasting, glucagon stimulates glycogenolysis, gluconeogenesis and inhibits lipogenesis. Hepatic response to insulin and glucagon is dysregulated in subjects with insulin resistance and type 2 diabetes [47]. Of note, a plethora of preclinical data and human studies suggests that abnormal accumulation of lipidic molecules and their byproducts such as triglycerides, ceramides, DAGs and long-chain fatty CoA molecules are mechanistically linked to the development of insulin resistance and non-alcoholic fatty liver disease (NAFLD) [48,49,50]. At the molecular level, lipid sensing in the liver requires a complex regulatory network. As mentioned above; this review describes a DAG-sensitive PKC and PKD isoforms and their role in the regulation of metabolism.
The PKC family is the most studied in the context of hepatic metabolism. PKCε was proposed as a DAG sensing kinase functionally linked to the insulin signaling in the liver. It was shown that the accumulation of sn-1,2-DAG activates PKCε. However, exact mechanisms leading PKCε activation in the liver are still under debate [51, 52]. Upon activation, PKCε induces phosphorylation of insulin receptor substrate 1 (IRS1) at Ser1101, which blocks insulin signaling. A number of studies using genetic approaches to suppress PKCε signaling by using antisense oligonucleotides or by the targeted deletion in the whole body of mice have shown that inactivation of PKCε protects against insulin resistance induced by short and long term high-fat diet feeding [49, 53, 54]. Nevertheless, Brandon and colleagues have shown that liver-specific inactivation of PKCε does not affect insulin signaling in this organ. By contrast, the specific inactivation of PKCε in the adipose tissue might be involved in crosstalk with the liver, which elicits changes at hepatic gene expression level affecting metabolic fitness in this organ [55]. In addition, further research has suggested that PKCε deletion in the liver promotes ketogenesis and paradoxically suppresses gluconeogenesis [56]. Therefore, the relevance of PKCε in hepatic insulin signaling must be interpreted cautiously. Although there is plenty of evidence that suggests a clear biological role of different lipid species in the development of insulin resistance and NAFLD, PKCε signaling in this context requires further investigation [57].
Despite the fact that PKCε is the best characterized PKC isoform in the liver, other isoforms also seem to play a role in the regulation of hepatic metabolism. PKCθ is highly induced in the hepatic cell line, HepG2, upon insulin and glucose stimulation, which correlates with the degradation of the IRS-1 leading to impaired insulin signaling. Moreover, genetic inactivation of PKCθ with siRNA ameliorates insulin resistance in cells [58]. Additionally, PKCθ is activated by Ca2+ signaling upon hypoxic stress in hepatic stellate cells, triggering autophagy [59]. However, the physiological contribution of PKCθ in vivo is still controversial as deletion of PKCθ in whole body of mice leads to higher susceptibility to develop obesity, insulin resistance and lower energy expenditure [60]. Therefore, to clearly delineate the function of PKCθ in the regulation of hepatic metabolism, the generation of animals carrying a liver-specific deletion of PKCθ would be required.
The novel PKCδ has also been linked to the onset of insulin resistance. Acute elevation of free fatty acids (FFAs) by an intra-venus infusion of lipids and heparin activates the axis of PKCδ – NADPH oxidase increasing oxidative stress which suppresses insulin signaling [61, 62]. In line with these findings, it has been proposed that PKCδ deletion in hepatocytes upregulates the antioxidant system in the liver [63]. Consistently, deletion of PKCδ in the whole body of mice revealed a reduced expression of key pro-apoptotic genes and caspase 9 in a model of nonalcoholic steatohepatitis induced by a choline-deficient diet (MCD). However, the authors did not find significant differences in pro-fibrotic genes expression after 8 weeks of MCD diet in PKCδ-deficient mice compared to the control littermates [64]. In the genetic models of obesity, mice carrying inactivation mutation in the leptin receptor (so-called db/db mice) and in Zucker rats, PKCδ deficiency prevents hepatic triglyceride accumulation [65, 66], promotes insulin signaling by restoring AKT as well as glycogen synthases kinase β (GSK3β) activity and induces glucose uptake [65, 67]. Moreover, increased genetic susceptibility to develop diabetes in C57BL/6 J compared to S129S6/Sv strain of mice [68] could be partially related to changes in the locus activity of the gene encoding PKCδ [69]. In line with these findings, liver-specific overexpression of PKCδ aggravates diabetes and promotes hepatosteatosis. Moreover, hepatic mRNA levels of PKCδ correlate positively with fasting glucose and circulating triglycerides in obese people [69].
Another essential hallmark in the onset of fatty liver disease is dysregulation of cholesterol metabolism. PKCβ is a major regulator of cholesterol and fatty acid metabolism in the liver [70]. High-fat diet and high cholesterol diets increase PKCβ expression [71, 72]. Mechanistically, PKCβ phosphorylates and controls the nuclear translocation of Farnesoid X receptor (FXR) a master regulator for cholesterol removal through its conversion into bile acids in the liver [73]. Conversely, full inactivation of PKCβ in mice promotes diet-induced gallstone disease [74]. On the other hand, PKCβ-deficient mice are protected against diet-induced obesity, insulin resistance and ectopic accumulation of fat in the liver [75]. Furthermore, it has been shown that PKCβ activation, both in vitro and in vivo with insulin sustains de novo lipogenesis by regulating the expression and activity of sterol regulatory element-binding protein 1c (SREBP-1c) an essential regulator of the lipogenic machinery [76].
PKD isoforms represent another group of DAG sensing kinases in hepatocytes. Moreover, PKC isoforms can activate PKDs in multiple cell types [77]. Recently, it was shown that out of three PKD isoforms described (PKD1, PKD2, PKD3) only PKD3 is significantly expressed in hepatocytes. PKD3 can be activated in hepatocytes in response to the elevation in DAG content in the hepatocytes and livers of mice fed HFD [14]. Moreover, it was demonstrated that PKD3 suppresses insulin signaling in mice fed HFD, but the exact molecular mechanisms need to be identified. Hence, mice with liver-specific deletion of PKD3 present better insulin sensitivity and consequently glucose handling [14]. However, activation of insulin signaling in mice deficient for PKD3 is associated with enhanced lipogenesis and consequently leads to the accumulation of triglycerides and cholesterol in the liver [14]. Interestingly, the deletion of PKD3 in immune cells promotes liver fibrosis by activating the production of transforming growth factor β (TGFβ), a classical pro-fibrotic cytokine, by hepatic macrophage [78]. On the other hand, the deletion of PKD1 specifically in adipocytes protects the development of od liver steatosis evoked by HFD feeding [79].
In conclusion, a number of PKC and PKD isoforms in the liver present a spectrum of non-redundant functions in the regulation of hepatic metabolism. Generally, PKCs and PKD3 block insulin signaling at different levels contributing to the development of insulin resistance and regulate hepatic lipogenesis contributing to the development of liver steatosis.
PKCs and PKDs regulate differentiation and function of adipocytes
Mammalian adipose tissue possesses a remarkable capacity to expand and to adapt to the fluctuations in nutrients supply. Thus, adipocytes have a major role in preventing the ectopic accumulation of fat in organs such as liver, skeletal muscle, heart, and pancreas [80]. Adipose tissue regulates energy storage, adaptive thermogenesis, endocrine function, food intake and provides fuels to peripheral organs. Adipocytes are classified into three types, based on their main features; white, beige, and brown adipocytes. Of note, beige adipocytes can emerge from white adipocytes or adipogenic precursors [81, 82]. Importantly, all of adipocytes types can store fat and produce adipokines, however, only beige and brown adipocytes can dissipate energy in the form of heat. For this reason, targeting beige and brown adipocytes represents a promising strategy to counteract obesity and type 2 diabetes.
Classical reports from the early 90s proposed a possible link between insulin signaling and PKCs in adipocytes and identified PKCβ, PKCγ and PKCε as the main PKC isoforms in adipocytes [83, 84]. Over the last two decades, significant advancements were made in the field of PKCs and their relevance in adipose tissue biology and obesity [85]. Importantly, the inactivation of PKCγ and PKCε impairs adipose differentiation in 3 T3-L1 cells by affecting the expression of PPARγ and CEBPα, essential adipogenic markers, while other PKC isoforms do not influence differentiation process [86]. However, other PKCs might influence other aspects of adipocytes biology. For instance, PKCβ deletion in the whole body of mice protects against diet-induced obesity. These animals present reduced adiposity, higher energy expenditure, increased expression of oxidative genes, improved mitochondrial fitness, higher levels of adrenergic receptors to sustain fat mobilization [87, 88]. Consistently, rodent models of obesity present higher levels of PKCβ in adipose tissue [87]. In line with this, human data have shown that activation of PKCβ by antipsychotic drugs promotes adiposity [89] and single nucleotide polymorphisms in the PKCβ promoter correlate negatively with insulin sensitivity [90]. Deletion of another isoform, PKCε, in adipose tissue results in protection from diet-induced insulin resistance and glucose intolerance [55]. Moreover, PKCε might promote glucose uptake in adipocytes while PKCθ inhibits adiponectin expression [91,92,93,94,95].
PKDs in adipocytes are activated by DAGs and extracellular purines [77]. Recently, it was demonstrated that PKD1 deletion in adipocytes protects against obesity and diabetes, by inducing thermogenic adipocytes (beige cells) and promoting the expression of genes activating energy dissipation by adipocytes such as uncoupling protein 1 (UCP1), PR-domain containing 16 (PRDM16) or peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) [79]. Moreover, the deletion of PKD1 in adipocytes promotes mitochondrial fragmentation and biogenesis. Finally, it was shown that PKD1 regulates adipocyte’s function by targeting and suppressing the activity of AMP-activated protein kinase (AMPK), as all the phenotypes observed in mice deficient for PKD1 in adipocytes were reversed by AMPK inactivation [79]. However, the impact of other PKD isoforms, PKD2 and PKD3 on adipose tissue function needs to be investigated in future.
Therefore, PKCs and PKDs regulate adipocytes’ acquisition and multiple aspects of their function.
PKCs and PKDs regulate insulin secretion in pancreatic β-cells
The main function of pancreatic β-cells found in the endocrine part of pancreas – pancreatic islets, is to produce and release insulin in response to the elevated glucose level in blood. Transport of glucose into β-cells is followed by the closing of the ATP-sensitive potassium channel and depolarization that leads to calcium influx via voltage-gated calcium channel [96]. Subsequently, the calcium influx activates PLC, which in turn hydrolases PIP2 and generates DAG [97]. Moreover, PLC in β-cells can also be activated through G-protein-coupled receptor pathways [98, 99]. An elevated level of DAG in β-cells causes activation of PKCs and PKDs and is associated with increased insulin secretion [96]. However, prolonged accumulation of intracellular lipids can be toxic, may promote β-cell failure, and contribute to type 2 diabetes development [100]. Moreover, sustained fatty acid overload results in the synthesis and accumulation of DAG in β-cells, which is correlated with impaired insulin secretion [101].
Several studies proved that treatment with an unspecific PKC activator, phorbol 12-myristate 13-acetate (PMA), stimulates insulin secretion in β-cells [102, 103]. PMA does not mediate intracellular calcium influx, nevertheless, it potentiates sensitivity of β-cells to calcium, decreasing the concentration of calcium needed to promote insulin exocytosis [103]. The effect of PMA was first linked to activation of PKCs, nevertheless, this agonist was also shown to activate PKDs [104]. PKCα, PKCβI, PKCβII, PKCδ, PKCθ, PKCη, and PKCε are expressed in pancreatic β-cells [105, 106]. Several studies showed that PKCs inhibitors strongly attenuate insulin secretion. Ro 31-8220, a non-selective PKC inhibitor partially reduced glucose-induced insulin secretion [107]. Moreover, Gö6976, an inhibitor of cPKCs significantly reduced the second phase of glucose-induced insulin secretion but not the initial one [108]. It was also observed that alike PMA, glucose and potassium-driven depolarization can cause translocation of PKCs to the plasma membrane [109,110,111,112,113,114]. Translocation dynamic upon DAG elevation in the plasma membrane differs depending on the isoform of PKC [105]. Interestingly, Yedovitzky et al. showed that glucose stimulation induces translocation of PKCα and PKCε to the periphery but PKCδ to the perinuclear site [110]. Furthermore, inhibition of PKCα and PKCε translocation reduces glucose-induced insulin release [110]. β-cells treatment with calcium-free buffer results in decreased translocation of PKCα, but not PKCε, and is associated with partially abolished insulin response [110]. Several studies showed the importance of PKCα in glucose and potassium-induced insulin granules secretion. However, Mendez et al. showed that after glucose treatment PKCε, but not PKCα, associates with insulin granules, which is essential for insulin exocytosis [112].
Overall, increased PKCs activity potentiates glucose-induced insulin secretion enhancing the second phase of insulin granule exocytosis. Based on the data obtained also from other cell types, it was proposed that PKCs regulate cortical actin rearrangement that facilitates insulin secretion and that the effect of PKCs is dependent on such exocytotic proteins as synaptotagmin, the mammalian homolog of UNC-18 (Munc18) and synaptosomal nerve-associated protein 25 (SNAP-25) [96]. However, the exact molecular mechanism of PKCs action on insulin release process in β-cells is not fully known yet and needs further studies. Pathways potentially involved in this phenomenon are discussed in detail elsewhere [96].
Additionally, PKCs are also involved in non-glucose-stimulated insulin release in β-cells. PKCs are shown to be effectors of glucagon-like peptide 1 (GLP-1) [115]. Shigeto et al. showed that GLP-1 activates PLC, which in turn leads to PKCs activation and subsequently membrane depolarization and insulin release [115]. The PKCs-mediated effect of GLP-1 is dependent on the activation of Na+-permeable transient receptor potential ion channels (TRPM) 4 and 5 [116]. It was also observed that PKCs inhibitors partially attenuate fatty-acid-induced enhancement of insulin secretion [117,118,119]. Moreover, using several inhibitors it was shown that nPKCs mediate the signal from non-nutrient secretagogues and enhance mitochondrial respiration and subsequently potentiate insulin release in INS1E cells and human pancreatic β-cells [120].
Several studies showed that fatty acid-treatment increases PKCs activity in β-cells [121,122,123]. Since elevated lipid level is known to induce β-cell dysfunction, PKCs were linked to the development of metabolic disorders [124]. Mice with global deletion of PKCε present enhanced insulin secretion upon fatty acids treatment and improved glucose tolerance when fed with HFD [124]. Genetic and functional ablation of PKCε results in an increased insulin secretion ex vivo in islets derived from diabetic mice deficient for the function of leptin receptor (db/db mice) which were pretreated with lipids. It was also observed that PKCɛ deletion restores the balance between lipid esterification and oxidation altered by fatty acid treatment in pancreatic islets [124]. Moreover, using PKCɛ-inhibitory peptide improves glucose-induced insulin release and glucose tolerance in diabetic db/db mice [124]. Similarly, overexpression of kinase-negative PKCδ in mice protects from HFD-induced glucose intolerance, increases insulin level and pancreatic islets size, and decreases apoptosis marker cleaved caspase-3 in β-cells in comparison to control animals [125]. Furthermore, overexpression of kinase-negative PKCδ protects isolated islets and INS1E cells from palmitic acid-induced apoptosis and mitochondrial dysfunction [125]. However, β-cell-specific overexpression of wild type PKCδ does not influence glucose tolerance, insulin plasma level or islet size in mice fed both, standard laboratory diet and HFD [126]. Moreover, insulin content and glucose-induced insulin secretion in INS1E cells overexpressing wild type PKCδ were similar to control, which shows that increased level of PKCδ did not impair β-cell function [126]. This data indicate that PKCδ and PKCε are important players in lipid-induced β-cell dysfunction and pathogenesis of type 2 diabetes, however PKCδ upregulation in β-cells is not sufficient to induce diabetic phenotype.
Streptozotocin treatment induces type 1 diabetes and this model is widely used to study autoimmune distruction of β-cells [127]. Global deletion of PKCδ delays the onset of hyperglycemia in streptozotocin-treated mice, confirming the importance of this PKC isoform in the pathogenesis of type 1 diabetes [128]. Furthermore, it was shown that the deletion of PKCδ in pancreatic islets protects from cytokine-induced apoptosis, NO generation, and also disrupts toll-like receptor 2 (TLR2) signaling, which activation contributes to the development of type 1 diabetes [128,129,130]. GLP-1 analog exendin-4 can suppress apoptosis and protects against oxidative damage in β-cells. By using specific inhibitor Kim et al. showed that the effect of exendin-4 on the expression of antioxidant genes mediated by nuclear factor erythroid 2-related factor 2 (Nrf2) in β-cells upon stress is dependent on PKCδ activity [130]. Apoptosis in β-cells is also regulated via transcription factor forkhead box protein O1 (FOXO1), that in dephosphorylated form increases expression of pro-apoptotic genes. It was shown that PKCδ regulates nuclear retention of FOXO1 in β-cells via phosphorylation of 14–3-3ζ chaperone [126]. Under non-stress conditions, both, INS1 overexpressing PKCδ and islets derived from mice with β cell-specific overexpression of PKCδ are characterized by increased accumulation of phosphorylated form of FOXO1 in the nucleus but do not display increased apoptosis [126]. Taken together, these data indicate an important role of PKCδ in the regulation of β-cell death and progression of type 1 diabetes development.
All the PKD isoforms are expressed in pancreatic islets, nevertheless, PKD1 is a dominant isoform [131,132,133]. PKD1 promotes granule secretion and fission of vesicles destined for exocytosis from the trans-Golgi network (TGN) [134]. It was shown that PKD1 plays an important role in the regulation of insulin secretion and the survival of β-cells in the pathogenesis of diabetes mellitus [132]. In β-cells, PKD1 activity is regulated at multiple levels. PKD1 is activated by the neurotransmitter derived from the parasympathetic nervous system, acetylcholine, and inhibited by mitogen-activated protein kinase (MAPK) p38δ (which directly phosphorylates PKD1) upon oxidative stress. Consistently, deletion of p38δ in mice leads to elevated PKD1 activity, enhanced insulin granule secretion, better glucose tolerance and protects against pancreatic β-cell failure [132]. Moreover, PKD1 is also a downstream effector of fatty acid receptor 1 (GPR40), which promotes the second phase of fatty acid-induced insulin release, and controls actin cytoskeleton remodeling in β-cells [135]. On the molecular level, PKD1 controls the biogenesis of insulin granules at the TGN by phosphorylating a BAR-domain-containing protein Arfaptin-1 at serine 132 [136]. Arfaptin-1 prevents the premature fission of the secretory granules from the TGN. PKD-dependent phosphorylation of Aprfaptin-1 allows fission of the insulin granules TGN and consequently its secretion [136].
Recently, PKDs were found to mediate adaptation to nutrient availability in β-cells. Upon feeding, PKDs block starvation-induced nascent granule degradation (SINGD), therefore promoting insulin granule secretion. De-activation of PKDs during nutrient deprivation allows the fusion of insulin granules with lysosomes and their subsequent degradation. This leads to the activation of the mechanistic target of rapamycin (mTOR) and subsequently autophagy suppression, which is required to inhibit insulin secretion during nutrient deprivation [137]. Another study showed that PKD prevents SINGD, partially in the CD63-dependent manner. Inhibition of PKD induces SINGD, which contributes to the β-cells failure in diabetic BTBRob/ob mice [131]. Mice carrying β-cell-specific inducible deletion of PKD1 present similar glucose tolerance and insulin levels as control animals when fed a standard diet. However, when fed with HFD mice with PKD1 deletion in β-cells were characterized by glucose intolerance and impaired glucose-induced insulin release [138]. This suggests that PKD1 is involved in β-cell adaptation to increased insulin demand upon HFD and protects against β-cell dysfunction [138].
**ao et al. for over 3 years studied a cohort of rhesus monkeys with spontaneous metabolic syndrome. They observed that monkeys with hyperinsulinemia, which is a condition implicated in the pathogenesis of insulin resistance and type 2 diabetes, were characterized by highly reduced expression of PKD2 due to nonsense mutation (K410X) [133]. It was also shown that at a young age global PKD2 knockout mice are characterized by increased fasting insulin level and decreased fasting glucose level in comparison to control animals [133]. Moreover, adult mice with PKD2 deficiency exhibit increased basal and glucose-stimulated insulin secretion, which is associated with augmented L-type calcium channel-dependent calcium influx in response to membrane depolarization induced by potassium chloride and glucose in those animals. Adult PKD2 knockout mice are also characterized by improved glucose tolerance and slightly decreased insulin tolerance [133].
Taken together, PKCs and PKDs play an important role in the regulation of insulin secretion and the survival of pancreatic β-cells.
PKCs suppress insulin sensitivity in skeletal muscle while PKDs are required for their function
Skeletal muscle is an organ that accounts for approximately 45% of the human body and is mainly responsible for body movement [139]. Since skeletal muscle is a highly metabolically active tissue responsible for up to 90% of glucose disposal in the postprandial state, it is considered to play a crucial role in maintaining whole-body energy homeostasis and development of metabolic disorders, for instance, insulin resistance and glucose intolerance [140]. Increased accumulation of DAG is inversely correlated with insulin sensitivity and is one of the factors causing the development of insulin resistance in skeletal muscle [141]. Moreover, elevated DAG level in muscles is associated with activation of PKCs and PKDs [142,143,144,145].
PKCα, PKCβ, PKCδ, PKCθ, and PKCε are expressed in skeletal muscle. However, PKCθ is recognized as a dominant isoform of DAG-sensing PKCs in this tissue [146]. Several studies have shown profoundly altered expression levels and activity of PKCs in skeletal muscle of obese human patients and rodent models of diabetes in comparison to healthy and lean controls [142, 143, 147,148,149]. In 1997, Bandyopadhyay et al. showed that PKCζ, but not DAG-dependent PKCs, is involved in insulin-stimulated glucose uptake in L6 myotubes in PI3K dependent manner [150]. However, Itani et al. observed increased translocation of PKCβ, PKCδ, and PKCθ from the cytosol to the membrane after insulin treatment in human skeletal muscle indicating its increased activation [148]. Moreover, insulin treatment causes alternative splicing of PKCβ pre-mRNA in favor of the PKCβII isoform and its activation is mediated by PI3K in skeletal muscle cells. In turn, activated PKCβII promotes glucose transportation via phosphorylation of its substrate myristoylated alanine-rich C-kinase substrate (MARCKS) [151].
Lack of PKCθ protects skeletal muscle from lipid-induced insulin resistance [152]. HFD-fed mice with skeletal muscle-specific PKCθ deficiency are characterized by decreased intracellular lipid accumulation and increased insulin sensitivity in skeletal muscle, decreased fasting glucose level, and lower daily calorie intake followed by lower weight gain in comparison to control mice [153]. Moreover, it was shown that activation of PKCθ induces inhibitory phosphorylation of IRSs at several residues, e.g. pSer1101 and pSer307, which is associated with disruption of insulin signaling pathway and decreased PI3K and AKT activity [154, 155]. Global deletion of PKCα increases insulin-stimulated glucose transport in skeletal muscle in mice whereas knockout of PKCβ increases both basal and insulin-stimulated glucose uptake in isolated soleus muscle [156, 157]. PKCε is also implicated in regulating skeletal muscle metabolism. Administration of PKCε abrogating peptides protect skeletal muscle from diet-induced insulin resistance and decreases phosphorylation of IRS-1 [158]. Interestingly, skeletal muscle-specific knockout of PKCδ improves age-related decline in whole-body insulin sensitivity and glucose tolerance, and also increases insulin sensitivity of skeletal muscle in older mice but does not protect from HFD-induced insulin resistance. Moreover, skeletal muscle with deficiency of PKCδ is characterized by decreased metabolic rate and lower level of OXPHOS proteins, indicating an important role of PKCδ in the regulation of mitochondria homeostasis at an older age [159]. The data published up to date indicate that all studied DAG-sensing PKCs regulate insulin pathways by providing a negative feedback loop, subsequently decreasing insulin-stimulated glucose uptake and other insulin-dependent processes in muscle cells. Overall, PKCs play a crucial role in DAG-induced insulin resistance in skeletal muscle.
Exercise is found as an effective treatment of insulin resistance in peripheral tissues. Rao et al. showed that physical training decreases the level of PKCβ but not of other PKC isoforms in skeletal muscle of HFD-fed mice. Knockout of PKCβ protects mice from HFD-induced insulin resistance in skeletal muscle in a similar way to exercise. Moreover, physical training does not influence skeletal muscle insulin sensitivity in PKCβ deficient mice fed with HFD. This data suggests that exercise protects against HFD-induced insulin resistance in skeletal muscle at least partially through the downregulation of PKCβ [160].
Moreover, it was shown that PKCθ regulates myoblast differentiation in C2C12 cells. PKCθ knockdown reduced inhibitory IRS-1 phosphorylation, increased extracellular signal-regulated kinase (ERK) 1/2 phosphorylation, enhanced myoblast differentiation and cell fusion, and increased protein synthesis [161]. Also, PKCε in skeletal muscle is involved in the differentiation of muscle cells and myofiber regeneration. During differentiation, PKCε localizes in the nucleus and blocks Hmga1 gene expression to promote myoblast formation [162].
In skeletal muscle PKCε and PKCβ1 are also involved in transmitter release at the neuromuscular junction [163, 164]. It was shown that both PKCε and PKCβ1 are localized in the motor nerve terminals of the neuromuscular junction. Expression and phosphorylation level of those PKCs in the synaptic membrane is increased in response to electrical stimulation and muscle contraction via the brain-derived neurotrophic factor (BDNF)-mediated tyrosine kinase receptor B (TrkB) signaling [163, 164]. Additionally, it was shown that muscle contraction regulates PKCβ1 phosphorylation in the synaptic membrane through phosphoinositide-dependent kinase 1 (PDK1) [164]. PKCε causes phosphorylation of MARCKS involved in neurotransmission-related actin cytoskeleton remodeling [163]. Using a specific inhibitor it was also shown that PKCε is involved in the regulation of acetylcholine release in the neuromuscular junction [163].
PKCs are involved in saturated fatty acid-induced proinflammatory signaling in skeletal muscle. Palmitic acid treatment increases intracellular DAG and subsequently induces PKC-dependent Ser153 phosphorylation of Raf-kinase inhibitor protein (RKIP) that results in RKIP-Raf1 dissociation and activation of ERK signaling pathway [165]. It is known that inflammation plays an important role in the pathogenesis and progression of skeletal muscle disorders, for instance, insulin resistance and Duchenne Muscular Dystrophy (DMD). DMD is a severe muscle disease caused by a mutation in the dystrophin gene. It was shown that knockout of PKCθ in the mouse model of DMD (mdx) prevents muscle inflammation, reduces muscle wasting, improves muscle regeneration and maintenance of performance [166]. Administration of PKCθ specific inhibitor to young mdx mice also leads to a reduction in muscle damage, immune cell infiltration, muscle inflammation, and maintenance of muscle regeneration [167]. These data suggest that specific PKCθ inhibitor may potentially be used in the treatment of DMD [167].
All of the PKD isoforms are expressed in skeletal muscle; however, the level of expression can differ between myofiber types [168, 169]. For instance, PKD1 is the most abundant isoform and is predominantly expressed in type I oxidative, slow-twitch myofibers. Moreover, it was shown that overexpression of constitutively active PKD1 in type II glycolytic, fast-twitch myofibers leads to transformation into type I myofibers phenotype [169]. Skeletal muscle consisted mostly of type I myofibers in mice with skeletal muscle-specific PKD1 knockout and are characterized by increased susceptibility to fatigue [169]. The data suggest that PKD1 regulates muscle fiber phenotype through phosphorylation of class II histone deacetylases (HDACs), a repressors of myocyte enhancer factor-2 (MEF2) transcription factor regulating the expression of genes involved in differentiation of myoblasts into multi-nucleated myotubes [169,170,171]. Moreover, it was shown that PKD1 mediates HDAC5 nuclear efflux as an effector of α-adrenergic signal, which is typically present in parallel with motor neuron input during physical training [172]. It was also shown that mice with overexpression of dominant-negative PKD1 exhibited decreases running performance and significantly impaired voluntary running-induced myofiber transformation in skeletal muscle, which confirms that PKD1 plays a crucial role in skeletal muscle remodeling upon physical training [173]. Coughlan et al. showed that PMA acting as DAG mimetic increases phosphorylation of AMPK, one of the major energy regulators in the cell, decreasing activity of its subunit AMPKα2 in the time and dose-dependent manner in C2C12 [144]. PKD1 specific inhibitor and PKD1 knockdown fully prevent PMA-induced effects on AMPK, whereas PKCs inhibitor acts only partially. Interestingly it was also shown that PKD1 inhibitor protected C2C12 cells from insulin resistance induced by PMA [144]. Moreover, PKD1 was found to directly phosphorylate AMPKα2 at Ser491 in in vitro studies [144]. These data suggest that PKD1 can be involved in insulin signaling impairment via AMPK inhibition.
Inhibition of PKDs decreases myotube fusion and myoblast differentiation in primary mouse satellite cells and C2C12 stable cell line [168]. Furthermore, it was shown that PKD2 is more relevant in the regulation of those processes than PKD1 and PKD3 isoforms. PKD2 is phosphorylated and, at the same time, activated during the initiation of C2C12 myoblasts differentiation, whereas knockdown of PKD2 leads to inhibition of myoblast cell fusion and impaired expression of muscle development-associated genes [168]. Kleger et al. observed that PKD2 activates the promyogenic transcription factor MEF2D via inhibition of paired box gene 3 (Pax3) [168]. Overexpression of dominant-negative PKD3 inhibits basal glucose transport but has only a minor effect on insulin-stimulated glucose uptake. Additionally, overexpression or silencing of PKD3 causes respectively, significant increase or decrease in basal glucose uptake in L6 myotubes [174]. Moreover, using a truncated form of the PKD catalytic domain, it was shown that PKDs also regulate expression of proteins involved in glucose and lipid metabolism, β-oxidation, and OXPHOS in C2C12 myotubes both in HDAC5-dependent and independent manner [175].
In conclusion, PKCs in skeletal muscle suppress insulin action while PKDs promotes muscle differentiation and function.
PKCs and PKDs in the regulation of appetite and food digestion
The regulation of food intake and nutrients absorption in the gastrointestinal tract determines glucose and lipid homeostasis. The hypothalamic arcuate nucleus plays a central role in the integration of hormonal and nutritional signals which regulate metabolic homeostasis [176]. Animals fed HFD present increased intracellular DAG levels in the hypothalamus [177]. Nevertheless, the impact of PKCs and PKDs on the hypothalamic regulation of metabolism is poorly understood. Administration of specific PKC agonists into the hypothalamus suppresses hepatic gluconeogenesis. Importantly, this effect can be reversed by PKCδ-specific inhibitor, rottlerin [178]. On the other hand, palmitic acid promotes the hypothalamic accumulation of DAG to stimulate the translocation of PKCθ to the plasma membrane. Importantly, PKCθ mediates palmitic acid-induced suppression of insulin signaling in the arcuate nucleus promoting body weight gain and glucose intolerance in mice fed HFD [179].
The gastrointestinal tract is the first side challenged by high-fat-containing foods. Therefore, it would be logical to assume that the fat challenge leads to the accumulation of DAG and subsequent activation of PKCs and PKDs in the intestine. However, up to date, there are no studies available that would cover this topic. Nevertheless, several studies implicated PKC and PKD isoforms in the regulation of intestinal epithelial cells proliferation and differentiation [180, 181].
PKCs and PKDs – beyond the regulation of glucose and lipid homeostasis
DAG evoked activation of PKCs and PKDs were also broadly discussed in the context of heart function. However, these aspects were recently revised in [182, 183]. Similarly, PKCs regulate multiple aspects of innate [184] and adaptive immunity [185], while PKDs plays a central role in the regulation of immune response [186, 187]. However, these aspects, likewise the PKCs and PKDs impact on other biological processes are beyond the scope of this review.
Targeting PKCs and PKDs – potential clinical applications
The PKCs and PKDs families play a pivotal role in metabolic regulation. Thus, targeting these kinases might represent a promising avenue for the treatment of metabolic disorders such as obesity and diabetes. Hitherto, several clinical trials have been carried out to target PKCs in the context of different clinical conditions. For metabolic research trials include diabetic retinopathy, congestive heart failure, coronary bypass grafting and acute myocardial infarction salvage [188,189,190,191,192,193,194,195,196]. Unfortunately, to date, due to limitations related to off-target effects, lack of specificity, lack of suitable metabolites in blood or urine to monitor the activity of the PKC inhibitors, as well as due to inconclusive preclinical studies, there is no commercially available drug on the market that targets specifically any of the isoforms of the PKCs [197]. Nevertheless, the pleiotropic effects of the PKCs make this family of kinases a potential target for the treatment of multiple metabolic diseases.
Concerning the PKD family there is no ongoing clinical trial register in clinicaltrial.gov. However, direct or indirect targeting of PKD isoforms might be an attractive strategy for preventing pancreatic β-cell failure during the onset of diabetes or for the treatment of obesity.
Conclusion and future perspective
In summary, this review provides comprehensive and updated insights into the classification, structure, tissue distribution and functions of the DAG-sensing PKCs and PKDs in health and metabolic diseases, with a major focus on organs involved in metabolic regulation, such as liver, adipose tissue, pancreas, and skeletal muscle. Besides, this review provides a brief overview of the current state of the art in PKCs and PKDs drug discovery and development. DAG-sensitive PKCs and PKDs play a crucial role in the regulation of metabolism in peripheral tissues. However, understanding of the complex interplay between different DAG-sensitive kinases and other components of signaling machinery requires further investigation. Moreover, targeting specific members of PKC and PKD families might be beneficial for the treatment of metabolic diseases especially type 2 diabetes and obesity. Nevertheless, highly specific inhibitors of selected PKCs and PKDs would be required to ameliorate the potential side effects of therapies against obesity, diabetes, and associated diseases.
Availability of data and materials
Not applicable.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- aPKC:
-
Atypical PKC
- ATGL:
-
Adipose triglyceride lipase
- cPKC:
-
Conventional PKC
- DAG:
-
Diacylglycerol
- DAGK:
-
Diacylglycerol kinase
- DGAT2:
-
Diglyceride acyltransferase
- DMD:
-
Duchenne muscular dystrophy
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FOXO1:
-
Forkhead box protein O1
- GLP-1:
-
Glucagon-like peptide 1
- HDACs:
-
Class II histone deacetylases
- HFD:
-
High-fat diet
- HSL:
-
Hormone-sensitive lipase
- IRS:
-
Insulin receptor substrate
- MARCKS:
-
Myristoylated alanine-rich C-kinase substrate
- MCD:
-
Choline-deficient diet
- Mdx:
-
Mouse model of Duchenne muscular dystrophy
- MEF2:
-
Myocyte enhancer factor-2
- NAFLD:
-
Non-alcoholic fatty liver disease
- nPKC:
-
Novel PKC
- PA:
-
Phosphatidic acid
- PIP2 :
-
Phosphatidylinositol 4,5-bisphosphate
- PKC:
-
Protein kinase C
- PKD:
-
Protein kinase D
- PL:
-
Phospholipid
- PLC:
-
Phospholipase C
- PMA:
-
Phorbol 12-myristate 13-acetate
- RKIP:
-
Raf-kinase inhibitor protein
- SINGD:
-
Starvation-induced nascent granule degradation
- TAG:
-
Triacylglycerol
References
Newton AC. Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol. 2018;53(2):208–30.
Cobbaut M, Van Lint J. Function and regulation of protein kinase D in oxidative stress: a tale of isoforms. Oxid Med Cell Longev. 2018;2018:2138502.
Eichmann TO, et al. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem. 2012;287(49):41446–57.
Coleman RA, Mashek DG. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev. 2011;111(10):6359–86.
Fagone P, Jackowski S. Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res. 2009;50(Supplement):S311–6.
Carrasco S, Mérida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32(1):27–36.
Eichmann TO, Lass A. DAG tales: the multiple faces of diacylglycerol—stereochemistry, metabolism, and signaling. Cell Mol Life Sci. 2015;72(20):3931–52.
Rodriguez JA, et al. In vitro stereoselective hydrolysis of diacylglycerols by hormone-sensitive lipase. Biochimica et Biophysica Acta (BBA)-Mol Cell Biol Lipids. 2010;1801(1):77–83.
Schweiger M, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem. 2006;281(52):40236–41.
Boni LT, Rando RR. The nature of protein kinase C activation by physically defined phospholipid vesicles and diacylglycerols. J Biol Chem. 1985;260(19):10819–25.
Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun. 1984;122(2):818–23.
Voelker DR, Kennedy EP. Cellular and enzymic synthesis of sphingomyelin. Biochemistry. 1982;21(11):2753–9.
Fukami K, et al. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog Lipid Res. 2010;49(4):429–37.
Mayer AE, et al. The kinase PKD3 provides negative feedback on cholesterol and triglyceride synthesis by suppressing insulin signaling. Sci Signal. 2019;12(593):eaav9150.
Schweiger M, et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat Commun. 2017;8(1):1–15.
Takai Y, et al. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem Biophys Res Commun. 1979;91(4):1218–24.
Silinsky EM, Searl TJ. Phorbol esters and neurotransmitter release: more than just protein kinase C? Br J Pharmacol. 2003;138(7):1191–201.
Brose N, Rosenmund C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115(23):4399–411.
Kazanietz MG. Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol Pharmacol. 2002;61(4):759–67.
Yang C, Kazanietz MG. Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol Sci. 2003;24(11):602–8.
Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308(5961):693–8.
Roose JP, et al. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol Cell Biol. 2005;25(11):4426–41.
Schmitz-Peiffer C, Biden TJ. Protein kinase C function in muscle, liver, and β-cells and its therapeutic implications for type 2 diabetes. Diabetes. 2008;57(7):1774–83.
Isakov N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin Cancer Biol. 2018;48:36–52.
Giorgione JR, et al. Increased membrane affinity of the C1 domain of protein kinase Cδ compensates for the lack of involvement of its C2 domain in membrane recruitment. J Biol Chem. 2006;281(3):1660–9.
Giorgione J, et al. Contribution of the C1A and C1B domains to the membrane interaction of protein kinase C. Biochemistry. 2003;42(38):11194–202.
Graybill C, et al. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J Biol Chem. 2012;287(25):21003–11.
Mérida I, et al. Diacylglycerol kinase control of protein kinase C. Biochem J. 2019;476(8):1205–19.
Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282(2):826–30.
Bollag GE, et al. Protein kinase C directly phosphorylates the insulin receptor in vitro and reduces its protein-tyrosine kinase activity. Proc Natl Acad Sci. 1986;83(16):5822–4.
Koshio O, Akanuma Y, Kasuga M. Identification of a phosphorylation site of the rat insulin receptor catalyzed by protein kinase C in an intact cell. FEBS Lett. 1989;254(1–2):22–4.
Lewis RE, et al. Threonine 1336 of the human insulin receptor is a major target for phosphorylation by protein kinase C. Biochemistry. 1990;29(7):1807–13.
Chin J, et al. Overexpression of protein kinase C isoenzymes alpha, beta I, gamma, and epsilon in cells overexpressing the insulin receptor. Effects on receptor phosphorylation and signaling. J Biol Chem. 1993;268(9):6338–47.
Chin JE, Liu F, Roth RA. Activation of protein kinase C alpha inhibits insulin-stimulated tyrosine phosphorylation of insulin receptor substrate-1. Mol Endocrinol. 1994;8(1):51–8.
Bossenmaier B, et al. Protein kinase C isoforms β 1 and β 2 inhibit the tyrosine kinase activity of the insulin receptor. Diabetologia. 1997;40(7):863–6.
Ellwanger K, Hausser A. Physiological functions of protein kinase D in vivo. IUBMB Life. 2013;65(2):98–107.
Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol. 2012;81(3):284–91.
Fu Y, Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep. 2011;12(8):785–96.
Chen J, et al. Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family. Biochem J. 2008;411(2):333–42.
Malhotra V, Campelo F. PKD regulates membrane fission to generate TGN to cell surface transport carriers. Cold Spring Harb Perspect Biol. 2011;3(2):a005280.
Sánchez-Ruiloba L, et al. Protein kinase D intracellular localization and activity control kinase D-interacting substrate of 220-kDa traffic through a postsynaptic density-95/discs large/zonula occludens-1-binding motif. J Biol Chem. 2006;281(27):18888–900.
Baron CL, Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science. 2002;295(5553):325–8.
Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280(14):13205–8.
Li J, et al. The role of protein kinase D in neurotensin secretion mediated by protein kinase C-α/−δ and rho/rho kinase. J Biol Chem. 2004;279(27):28466–74.
Jacamo R, et al. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J Biol Chem. 2008;283(19):12877–87.
Sinnett-Smith J, et al. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J Biol Chem. 2009;284(20):13434–45.
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–71.
Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572.
Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38(7):649–65.
Ter Horst KW, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19(10):1997–2004.
Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol. 2018;19(10):654–72.
Petersen MC, et al. Insulin receptor Thr 1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126(11):4361–71.
Samuel VT, et al. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117(3):739–45.
Raddatz K, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia. 2011;54(6):1447–56.
Brandon AE, et al. Protein kinase C epsilon deletion in adipose tissue, but not in liver, improves glucose tolerance. Cell Metab. 2019;29(1):183–191. e7.
Raddatz K, et al. Deletion of protein kinase Cε in mice has limited effects on liver metabolite levels but alters fasting ketogenesis and gluconeogenesis. Diabetologia. 2012;55(10):2789–93.
Schmitz-Peiffer C. Deconstructing the role of PKC epsilon in glucose homeostasis. Trends Endocrinol Metab. 2020;31(5):344–56.
Haasch D, et al. PKCθ is a key player in the development of insulin resistance. Biochem Biophys Res Commun. 2006;343(2):361–8.
** Y, et al. Activation of autophagy through calcium-dependent AMPK/mTOR and PKC θ pathway causes activation of rat hepatic stellate cells under hypoxic stress. FEBS Lett. 2016;590(5):672–82.
Gao Z, et al. Inactivation of PKCθ leads to increased susceptibility to obesity and dietary insulin resistance in mice. Am J Physiol-Endocrinol Metab. 2007;292(1):E84–91.
Pereira S, et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am J Physiol-Endocrinol Metab. 2014;307(1):E34–46.
Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–8.
Mai HN, et al. Protein kinase Cδ knockout mice are protected from cocaine-induced hepatotoxicity. Chem Biol Interact. 2019;297:95–108.
Greene MW, et al. Lipid metabolism, oxidative stress and cell death are regulated by PKC delta in a dietary model of nonalcoholic steatohepatitis. PLoS One. 2014;9(1):e85848.
Zhang J, et al. PKCδ regulates hepatic triglyceride accumulation and insulin signaling in Leprdb/db mice. Biochem Biophys Res Commun. 2014;450(4):1619–25.
Greene MW, et al. PKCδ is activated in the liver of obese Zucker rats and mediates diet-induced whole body insulin resistance and hepatocyte cellular insulin resistance. J Nutr Biochem. 2014;25(3):281–8.
Brutman-Barazani T, et al. Protein kinase Cδ but not PKCα is involved in insulin-induced glucose metabolism in hepatocytes. J Cell Biochem. 2012;113(6):2064–76.
Haluzik M, et al. Genetic background (C57BL/6J versus FVB/N) strongly influences the severity of diabetes and insulin resistance in Ob/Ob mice. Endocrinology. 2004;145(7):3258–64.
Bezy O, et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J Clin Invest. 2011;121(6):2504–17.
Mehta D, Mehta KD. PKCβ: expanding role in hepatic adaptation of cholesterol homeostasis to dietary fat/cholesterol. Am J Physiol-Gastrointestinal Liver Physiol. 2017;312(3):G266–73.
Armstrong D, Zidovetzki R. Amplification of diacylglycerol activation of protein kinase C by cholesterol. Biophys J. 2008;94(12):4700–10.
Haeffner EW, Wittmann U. Free cholesterol induces activation but not translocation of protein kinase C in cultured ascites tumour cells. Cell Signal. 1994;6(2):201–7.
Gineste R, et al. Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol Endocrinol. 2008;22(11):2433–47.
Huang W, et al. Disruption of the murine protein kinase Cβ gene promotes gallstone formation and alters biliary lipid and hepatic cholesterol metabolism. J Biol Chem. 2011;286(26):22795–805.
Huang W, et al. Loss of protein kinase Cβ function protects mice against diet-induced obesity and development of hepatic steatosis and insulin resistance. Hepatology. 2009;49(5):1525–36.
Yamamoto T, et al. Protein kinase Cbeta mediates hepatic induction of sterol-regulatory element binding protein-1c by insulin. J Lipid Res. 2010;51(7):1859–70.
Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology. 2011;26(1):23–33.
Zhang S, et al. Deletion of protein kinase D3 promotes liver fibrosis in mice. Hepatology. 2020; https://doi.org/10.1002/hep.31176.
Löffler MC, et al. Protein kinase D1 deletion in adipocytes enhances energy dissipation and protects against adiposity. EMBO J. 2018;37(22):e99182.
Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–58.
Hepler C, Vishvanath L, Gupta RK. Sorting out adipocyte precursors and their role in physiology and disease. Genes Dev. 2017;31(2):127–40.
Carobbio S, et al. Brown and beige fat: from molecules to physiology and pathophysiology. Biochimica et Biophysica Acta (BBA)-Mol Cell Biol Lipids. 2019;1864(1):37–50.
Farese R, et al. Effects of insulin and phorbol esters on subcellular distribution of protein kinase C isoforms in rat adipocytes. Biochem J. 1992;288(1):319–23.
Frevert EU, Kahn BB. Protein kinase C isoforms ɛ, η, δ and ζ in murine adipocytes: expression, subcellular localization and tissue-specific regulation in insulin-resistant states. Biochem J. 1996;316(3):865–71.
Mehta KD. Emerging role of protein kinase C in energy homeostasis: a brief overview. World J Diabetes. 2014;5(3):385.
Fleming, I., et al., Protein kinase C isoforms play differential roles in the regulation of adipocyte differentiation. Biochem J, 1998. 333(3): p. 719–727.
Huang W, et al. Protein kinase Cβ deficiency attenuates obesity syndrome of Ob/Ob mice by promoting white adipose tissue remodeling. J Lipid Res. 2012;53(3):368–78.
Bansode RR, et al. Protein kinase Cβ deficiency increases fatty acid oxidation and reduces fat storage. J Biol Chem. 2008;283(1):231–6.
Pavan C, et al. Weight gain related to treatment with atypical antipsychotics is due to activation of PKC-β. Pharmacogenomics J. 2010;10(5):408–17.
Osterhoff MA, et al. Identification of a functional protein kinase Cβ promoter polymorphism in humans related to insulin resistance. Mol Genet Metab. 2008;93(2):210–5.
Lorenzo M, et al. PLCγ participates in insulin stimulation of glucose uptake through activation of PKCζ in brown adipocytes. Exp Cell Res. 2002;278(2):146–57.
Tsuchiya A, Kanno T, Nishizaki T. Diacylglycerol promotes GLUT4 translocation to the cell surface in a PKCε-dependent and PKCλ/ι and-ζ-independent manner. Life Sci. 2013;93(5–6):240–6.
Kotani K, et al. Requirement of atypical protein kinase Cλ for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18(12):6971–82.
Bosch RR, et al. Regulation of GLUT1-mediated glucose uptake by PKCλ–PKCβII interactions in 3T3-L1 adipocytes. Biochem J. 2004;384(2):349–55.
Sun S, et al. The inhibitory effects of PKCθ on adiponectin expression is mediated by ERK in 3T3-L1 adipocytes. J Endocrinol Invest. 2011;34(1):8–15.
Trexler AJ, Taraska JW. Regulation of insulin exocytosis by calcium-dependent protein kinase C in beta cells. Cell Calcium. 2017;67:1–10.
Thore S, et al. Feedback activation of phospholipase C via intracellular mobilization and store-operated influx of Ca2+ in insulin-secreting β-cells. J Cell Sci. 2005;118(19):4463–71.
Wuttke A, Idevall-Hagren O, Tengholm A. P2Y1 receptor-dependent diacylglycerol signaling microdomains in β cells promote insulin secretion. FASEB J. 2013;27(4):1610–20.
Henquin J-C. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52(5):739.
Janikiewicz J, et al. Islet β-cell failure in type 2 diabetes–within the network of toxic lipids. Biochem Biophys Res Commun. 2015;460(3):491–6.
Watson ML, et al. Chronic effects of palmitate overload on nutrientinduced insulin secretion and autocrine signalling in pancreatic MIN6 beta cells. PLoS One. 2011;6(10):e25975.
Arkhammar P, et al. Effects of protein kinase C activation on the regulation of the stimulus-secretion coupling in pancreatic β-cells. Biochem J. 1989;264(1):207–15.
Ammälä C, et al. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc Natl Acad Sci. 1994;91(10):4343–7.
Guo J, et al. Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem. 2011;286(8):6500–9.
Wuttke A, Yu Q, Tengholm A. Autocrine signaling underlies fast repetitive plasma membrane translocation of conventional and novel protein kinase C isoforms in β cells. J Biol Chem. 2016;291(29):14986–95.
Tian Y-M, Urquidi V, Ashcroft S. Protein kinase C in beta-cells: expression of multiple isoforms and involvement in cholinergic stimulation of insulin secretion. Mol Cell Endocrinol. 1996;119(2):185–93.
Harris TE, Persaud SJ, Jones PM. Atypical isoforms of PKC and insulin secretion from pancreatic β-cells: evidence using Gö 6976 and Ro 31-8220 as PKC inhibitors. Biochem Biophys Res Commun. 1996;227(3):672–6.
Zawalich WS, Zawalich KC. Effects of protein kinase C inhibitors on insulin secretory responses from rodent pancreatic islets. Mol Cell Endocrinol. 2001;177(1–2):95–105.
Ganesan S, et al. Immunocytochemical localization of alpha-protein kinase C in rat pancreatic beta-cells during glucose-induced insulin secretion. J Cell Biol. 1992;119(2):313–24.
Yedovitzky M, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J Biol Chem. 1997;272(3):1417–20.
Mogami H, et al. Decoding of short-lived Ca2+ influx signals into long term substrate phosphorylation through activation of two distinct classes of protein kinase C. J Biol Chem. 2003;278(11):9896–904.
Mendez CF, et al. Rapid association of protein kinase C-ϵ with insulin granules is essential for insulin exocytosis. J Biol Chem. 2003;278(45):44753–7.
Pinton P, et al. Dynamics of glucose-induced membrane recruitment of protein kinase C βII in living pancreatic islet β-cells. J Biol Chem. 2002;277(40):37702–10.
Zhang H, et al. Bimodal role of conventional protein kinase C in insulin secretion from rat pancreatic β cells. J Physiol. 2004;561(1):133–47.
Shigeto M, et al. A role of PLC/PKC-dependent pathway in GLP-1-stimulated insulin secretion. J Mol Med. 2017;95(4):361–8.
Shigeto M, et al. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J Clin Invest. 2015;125(12):4714–28.
Warnotte C, et al. Mechanisms of the stimulation of insulin release by saturated fatty acids: a study of palmitate effects in mouse β-cells. Diabetes. 1994;43(5):703–11.
Littman ED, et al. Role of protein kinase C isoenzymes in fatty acid stimulation of insulin secretion. Pancreas. 2000;20(3):256–63.
Thams P, Capito K. Differential mechanisms of glucose and palmitate in augmentation of insulin secretion in mouse pancreatic islets. Diabetologia. 2001;44(6):738–46.
Santo-Domingo J, et al. Coordinated activation of mitochondrial respiration and exocytosis mediated by PKC signaling in pancreatic β cells. FASEB J. 2017;31(3):1028–45.
Alcáazar O, et al. Stimulation of islet protein kinase C translocation by palmitate requires metabolism of the fatty acid. Diabetes. 1997;46(7):1153–8.
Eitel K, et al. Protein kinase C δ activation and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes. 2003;52(4):991–7.
Wrede C, et al. Fatty acid and phorbol ester-mediated interference of mitogenic signaling via novel protein kinase C isoforms in pancreatic beta-cells (INS-1). J Mol Endocrinol. 2003;30(3):271–86.
Schmitz-Peiffer C, et al. Inhibition of PKCɛ improves glucose-stimulated insulin secretion and reduces insulin clearance. Cell Metab. 2007;6(4):320–8.
Hennige AM, et al. Overexpression of kinase-negative protein kinase Cδ in pancreatic β-cells protects mice from diet-induced glucose intolerance and β-cell dysfunction. Diabetes. 2010;59(1):119–27.
Gerst F, et al. Protein kinase Cδ regulates nuclear export of FOXO1 through phosphorylation of the chaperone 14-3-3ζ. Diabetologia. 2015;58(12):2819–31.
Wu J, Yan L-J. Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity. Diab Metab Syndrome Obes Targets Therapy. 2015;8:181.
Cantley J, et al. Deletion of protein kinase Cδ in mice modulates stability of inflammatory genes and protects against cytokine-stimulated beta cell death in vitro and in vivo. Diabetologia. 2011;54(2):380–9.
Carpenter L, Cordery D, Biden TJ. Protein kinase Cdelta activation by interleukin-1beta stabilizes inducible nitric-oxide synthase mRNA in pancreatic beta-cells. J Biol Chem. 2001;276(7):5368–74.
Kim M-H, et al. EX4 stabilizes and activates Nrf2 via PKCδ, contributing to the prevention of oxidative stress-induced pancreatic beta cell damage. Toxicol Appl Pharmacol. 2017;315:60–9.
Pasquier A, et al. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat Commun. 2019;10(1):1–14.
Sumara G, et al. Regulation of PKD by the MAPK p38δ in insulin secretion and glucose homeostasis. Cell. 2009;136(2):235–48.
**ao Y, et al. Deficiency of PRKD2 triggers hyperinsulinemia and metabolic disorders. Nat Commun. 2018;9(1):1–11.
Bard F, Malhotra V. The formation of TGN-to-plasma-membrane transport carriers. Annu Rev Cell Dev Biol. 2006;22:439–55.
Ferdaoussi M, et al. G protein-coupled receptor (GPR) 40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia. 2012;55(10):2682–92.
Gehart H, et al. The BAR domain protein Arfaptin-1 controls secretory granule biogenesis at the trans-Golgi network. Dev Cell. 2012;23(4):756–68.
Goginashvili A, et al. Insulin secretory granules control autophagy in pancreatic β cells. Science. 2015;347(6224):878–82.
Bergeron V, et al. Deletion of protein kinase D1 in pancreatic β-cells impairs insulin secretion in high-fat diet–fed mice. Diabetes. 2018;67(1):71–7.
Brown S, Sewry C. Basics of skeletal muscle function and Normal physiology. In: Cardioskeletal myopathies in children and Young adults. Elsevier; 2017. p. 21–38.
DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(suppl 2):S157–63.
Montell E, et al. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am J Physiol-Endocrinol Metab. 2001;280(2):E229–37.
Ikeda Y, et al. Cellular mechanism of nutritionally induced insulin resistance in Psammomys obesus: overexpression of protein kinase Cε in skeletal muscle precedes the onset of hyperinsulinemia and hyperglycemia. Diabetes. 2001;50(3):584–92.
Itani SI, et al. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51(7):2005–11.
Coughlan KA, et al. PKD1 inhibits AMPKα2 through phosphorylation of serine 491 and impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2016;291(11):5664–75.
Szendroedi J, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci. 2014;111(26):9597–602.
Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37.
Cooper DR, Watson JE, Dao M. Decreased expression of protein kinase-C alpha, beta, and epsilon in soleus muscle of Zucker obese (fa/fa) rats. Endocrinology. 1993;133(5):2241–7.
Itani SI, et al. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes. 2000;49(8):1353–8.
Schmitz-Peiffer C, et al. Alterations in the expression and cellular localization of protein kinase C isozymes ε and θ are associated with insulin resistance in skeletal muscle of the high-fat–fed rat. Diabetes. 1997;46(2):169–78.
Bandyopadhyay G, et al. Evidence for involvement of protein kinase C (PKC)-ζ and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology. 1997;138(11):4721–31.
Chappell D, et al. Functional involvement of protein kinase C-βII and its substrate, myristoylated alanine-rich C-kinase substrate (MARCKS), in insulin-stimulated glucose transport in L6 rat skeletal muscle cells. Diabetologia. 2009;52(5):901–11.
Kim JK, et al. PKC-θ knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114(6):823–7.
Peck B, et al. Mice lacking PKC-θ in skeletal muscle have reduced intramyocellular lipid accumulation and increased insulin responsiveness in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2018;314(3):R468–77.
Yu C, et al. Mechanism by which fatty acids inhibit insulin activation of IRS-1 associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277(52):50230–6.
Li Y, et al. Protein kinase C θ inhibits insulin signaling by phosphorylating IRS1 at Ser1101. J Biol Chem. 2004;279(44):45304–7.
Letiges M, et al. Knockout of PKCα enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16(4):847–58.
Standaert ML, et al. Effects of knockout of the protein kinase C β gene on glucose transport and glucose homeostasis. Endocrinology. 1999;140(10):4470–7.
Mack E, et al. Prevention of insulin resistance and beta-cell loss by abrogating PKCε-induced serine phosphorylation of muscle IRS-1 in Psammomys obesus. Diabetes Metab Res Rev. 2008;24(7):577–84.
Li M, et al. Role of PKCδ in insulin sensitivity and skeletal muscle metabolism. Diabetes. 2015;64(12):4023–32.
Rao X, et al. Exercise protects against diet-induced insulin resistance through downregulation of protein kinase Cβ in mice. PLoS One. 2013;8(12):e81364.
Marino JS, et al. Suppression of protein kinase C theta contributes to enhanced myogenesis in vitro via IRS1 and ERK1/2 phosphorylation. BMC Cell Biol. 2013;14(1):39.
Di Marcantonio D, et al. PKCε as a novel promoter of skeletal muscle differentiation and regeneration. Exp Cell Res. 2015;339(1):10–9.
Obis T, et al. The novel protein kinase C epsilon isoform modulates acetylcholine release in the rat neuromuscular junction. Mol Brain. 2015;8(1):80.
Hurtado E, et al. Synaptic activity and muscle contraction increases PDK1 and PKCβI phosphorylation in the presynaptic membrane of the neuromuscular junction. Front Mol Neurosci. 2017;10:270.
Macrae K, et al. Defining the role of DAG, mitochondrial function, and lipid deposition in palmitate-induced proinflammatory signaling and its counter-modulation by palmitoleate. J Lipid Res. 2013;54(9):2366–78.
Madaro L, et al. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS One. 2012;7(2):e31515.
Marrocco V, et al. Pharmacological inhibition of PKCθ counteracts muscle disease in a mouse model of Duchenne muscular dystrophy. EBioMedicine. 2017;16:150–61.
Kleger A, et al. Protein kinase D2 is an essential regulator of murine myoblast differentiation. PLoS One. 2011;6(1):e14599.
Kim M-S, et al. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol. 2008;28(11):3600–9.
Lu J, et al. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6(2):233–44.
McKinsey TA, et al. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408(6808):106–11.
Liu Y, et al. α-Adrenergic signalling activates protein kinase D and causes nuclear efflux of the transcriptional repressor HDAC5 in cultured adult mouse soleus skeletal muscle fibres. J Physiol. 2009;587(5):1101–15.
Ellwanger K, et al. Protein kinase D controls voluntary-running-induced skeletal muscle remodelling. Biochem J. 2011;440(3):327–35.
Chen J, Lu G, Wang QJ. Protein kinase C-independent effects of protein kinase D3 in glucose transport in L6 myotubes. Mol Pharmacol. 2005;67(1):152–62.
McGee SL, et al. Compensatory regulation of HDAC5 in muscle maintains metabolic adaptive responses and metabolism in response to energetic stress. FASEB J. 2014;28(8):3384–95.
Timper K, Brüning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech. 2017;10(6):679–89.
Borg ML, et al. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J Physiol. 2012;590(17):4377–89.
Ross R, et al. Hypothalamic protein kinase C regulates glucose production. Diabetes. 2008;57(8):2061–5.
Benoit SC, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. J Clin Invest. 2009;119(9):2577–89.
Wang J, et al. Positive cross talk between protein kinase D and β-catenin in intestinal epithelial cells: impact on β-catenin nuclear localization and phosphorylation at Ser552. Am J Physiol Cell Physiol. 2016;310(7):C542–57.
Frey MR, et al. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol. 2000;151(4):763–78.
Marrocco V, et al. PKC and PKN in heart disease. J Mol Cell Cardiol. 2019;128:212–26.
Simsek Papur O, et al. Acute and chronic effects of protein kinase-D signaling on cardiac energy metabolism. Front Cardiovasc Med. 2018;5:65.
Lim PS, Sutton CR, Rao S. Protein kinase C in the immune system: from signalling to chromatin regulation. Immunology. 2015;146(4):508–22.
Long A, Freeley M. Protein kinase C: a regulator of cytoskeleton remodelling and T-cell migration. Biochem Soc Trans. 2014;42(6):1490–7.
Ittner A, et al. Regulation of PTEN activity by p38δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med. 2012;209(12):2229–46.
Zhang Z, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med. 2017;214(9):2671–93.
Packer M, et al. Double-blind, placebo-controlled study of the efficacy of flosequinan in patients with chronic heart failure. J Am Coll Cardiol. 1993;22(1):65–72.
Association, A.D. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the protein kinase C β inhibitor diabetic retinopathy study (PKC-DRS) multicenter randomized clinical trial. Diabetes. 2005;54(7):2188–97.
Guarracino F, et al. Myocardial damage prevented by volatile anesthetics: a multicenter randomized controlled study. J Cardiothorac Vasc Anesth. 2006;20(4):477–83.
Tritapepe L, et al. Cardiac protection by volatile anaesthetics: a multicentre randomized controlled study in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. Eur J Anaesthesiol. 2007;24(4):323–31.
** Z, et al. The myocardial protective effects of adenosine pretreatment in children undergoing cardiac surgery: a randomized controlled clinical trial. Eur J Cardiothorac Surg. 2011;39(5):e90–6.
Mentzer RM Jr, et al. Adenosine myocardial protection: preliminary results of a phase II clinical trial. Ann Surg. 1999;229(5):643.
Mangano DT, et al. Post-reperfusion myocardial infarction: long-term survival improvement using adenosine regulation with acadesine. J Am Coll Cardiol. 2006;48(1):206–14.
Ross AM, et al. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol. 2005;45(11):1775–80.
Lincoff AM, et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: results of the PROTECTION AMI randomized controlled trial. Eur Heart J. 2014;35(37):2516–23.
Singh RM, et al. Protein kinase C and cardiac dysfunction: a review. Heart Fail Rev. 2017;22(6):843–59.
Acknowledgements
Authors would like to acknowledge Servier Medical Art by Servier (smart.servier.com) for sharing free images under a Creative Commons Attribution 3.0 Unported License. Some elements from smart.servier.com were used for creating the figures.
Funding
The authors of this review are funded by European Research Council (ERC) Starting Grant (SicMetabol, no. 678119), EMBO Installation Grant from European Molecular Biology Organization (EMBO), and the Dioscuri Centre of Scientific Excellence – The program initiated by the Max Planck Society (MPG), managed jointly with the National Science Centre and mutually funded by the Ministry of Science and Higher Education (MNiSW) and the German Federal Ministry of Education and Re-search (BMBF).
Author information
Authors and Affiliations
Contributions
KK wrote ‘Diacylglycerol (DAG) the structure and sources’, ‘PKC isoforms – structure and classification’, ‘PKDs – downstream effectors of PKCs and DAG’, ‘PKCs and PKDs regulate insulin secretion in pancreatic β-cells’, ‘PKCs suppress insulin sensitivity in skeletal muscle while PKDs are required for their function’, prepared Fig. 1 and partially Fig. 2. ALV wrote ‘DAG-evoked activation of PKCs and PKDs suppresses insulin signaling and hepatocytes’ and ‘PKCs and PKDs regulate differentiation and function of adipocytes’. IH wrote ‘PKCs and PKDs in the regulation of apetite and food digestion’ and partially prepared Fig. 2. GS wrote ‘PKCs and PKDs – beyond the regulation of glucose and lipid homeostasis’, ‘Targeting PKCs and PKDs: potential clinical applications’ and ‘Conclusion and future perspective’, coordinated the work, and corrected the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kolczynska, K., Loza-Valdes, A., Hawro, I. et al. Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: a review. Lipids Health Dis 19, 113 (2020). https://doi.org/10.1186/s12944-020-01286-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01286-8