
Abstract
Increasing evidence indicates that the ability of cancer cells to convey biological information to recipient cells within the tumor microenvironment (TME) is crucial for tumor progression. Microvesicles (MVs) are heterogenous vesicles formed by budding of the cellular membrane, which are secreted in larger amounts by cancer cells than normal cells. Recently, several reports have also disclosed that MVs function as important mediators of intercellular communication between cancerous and stromal cells within the TME, orchestrating complex pathophysiological processes. Chemokines are a family of small inflammatory cytokines that are able to induce chemotaxis in responsive cells. MVs which selective incorporate chemokines as their molecular cargos may play important regulatory roles in oncogenic processes including tumor proliferation, apoptosis, angiogenesis, metastasis, chemoresistance and immunomodulation, et al. Therefore, it is important to explore the association of MVs and chemokines in TME, identify the potential prognostic marker of tumor, and develop more effective treatment strategies. Here we review the relevant literature regarding the role of MVs and chemokines in TME.
Similar content being viewed by others


Background
Cells generate extracellular vesicles (EVs) which are small lipid membrane-enclosed particles and function as pivotal mediators of intercellular communication by transporting biological information between cells and their microenvironment [1]. Many cell types, ranging from embryonic stem (ES) cells [2, 3] to highly malignant cancer cells [4,5,6], are capable of releasing different classes of EVs. In terms of pathophysiological processes, EVs have been established as important players contributing to the development and progression of cancer, and are of relevance to diseases of various sorts [7,8,9,10], including autoimmune, inflammatory, cardiovascular, hematologic, and other diseases. Two main types of EVs have been described as exosomes and microvesicles (MVs) [1, 11]. In addition, recent data have demonstrated the existence of additional varieties of EVs, which may differ in size, biogenesis, and molecular cargo profiles [12].
Chemokines are a superfamily of small, chemoattractant cytokines that bind to and activate a family of the G-protein-coupled cell-surface receptors [13]. In cancer, chemokines and their receptors are important regulators for cell trafficking in and out of the tumor microenvironment (TME) [14]. In the TME, cancer cells and surrounding non-cancerous cells constantly exchange information via gap junctions, tunneling nanotubes and effector molecules. Membrane-enclosed EVs is one of the important cargos to ensure coordinated release of multiple molecules by packaging them together [15].
The biogenesis of MVs and chemokines
MVs, also commonly referred to as ectosomes or microparticles, are significantly larger in size than exosomes (100–1000 μm in diameter) [6, 16, 17] (Fig. 1). Unlike exosomes, the release of MVs typically involves centrifugal budding in specific areas of the plasma membrane [18]. Upon the release of Ca2+ from the endoplasmic reticulum, the plasma membrane undergoes molecular rearrangement at the sites where MVs originate, followed by direct shedding and instantaneous release of the vesicle into the intercellular space [10, 19]. MVs contain parental intracellular information and inherit partial cell membrane markers from which they are generated. Several proteins have been proposed MVs-specific, including selectins, integrins, CD40, matrix metalloproteinase (MMP), phosphatidylserine (PS), ADP-ribosylation factor 6 (ARF6) and Rho family members [11, 20]. Different types of MVs can form in various physiological and pathological conditions. Apoptotic blebs, for instance, are microvesicles released by cells upon the trigger of the cellular collapse that results in fragmentation of nucleus, increase in permeability of the plasma membrane, and externalization of PS [21]. During apoptosis, cellular components enclosed by apoptotic blebs are actively transferred from the apoptotic cell into peripheral vesicles [22]. Another example is the recently identified cancer-derived EV population, often termed as “large oncosome”, which is considerably larger than most known EV types characterized to date [11]. Biogenesis of large oncosomes is particularly notable in tumor cells with an amoeboid phenotype, which tend to be more aggressive. Similar to MVs, this EV population might originate directly from plasma membrane budding and, similar to MVs, these particles express ARF6 [23, 24].

Schematic structure model of microvesicle. ARF6: ADP-ribosylation factor 6, CD40: cluster of differentiation 40, EGFR: epidermal growth factor receptor, IL-1β: interleukin-1β, IL-6: interleukin-6, MMP: matrix metalloproteinase, tTG: tissue transglutaminase, uPA: urokinase plasminogen activator, VAMP-3: vesicle-associated membrane protein 3, VEGF: vascular epithelium growth factor, v-SNARE: vesicular soluble N-ethylmaleimide-sensitive factor attachment protein receptor
Chemokines are small proteins that act by combining with their cell surface receptors. They take active roles in numerous pathological states and biological processes, including immune response [25], tissue injury [26], cardiovascular diseases [27], and oncogenesis [28]. To date, more than 20 corresponding human chemokine receptors have been identified. Chemokines can be categorized by the position of the conserved cysteine residues [13, 29] into four different groups: C, CC, CXC, and CX3C. A chemokine receptor comprises seven-transmembrane domains, all with three extracellular and three intracellular loops. One of the intracellular loops is coupled with heterotrimeric G-proteins, capable upon binding of the ligand and receptor of triggering a cascade of signal transduction events [29,30,31]. Receptor nomenclature typically follows that of the chemokines, i.e. CC chemokines bind to CC chemokine receptors, CXC ligands bind to CXC receptors, with a high degree of redundancy in the chemokine family as multiple chemokines bind to the same receptor [29, 32] (Fig. 2).

Components of the chemokine systems. The majority of chemokines can bind a series of cognate receptors, and a single receptor can bind multiple chemokines, as exhibited in this paradigm for most CC (blue) and CXC (dark blue) chemokines. Atypical receptors (green) can also interact with numerous chemokines. On the contrary, a minority of receptors (red) has only one ligand
Tumor cells have the potential to sabotage the chemokine system, in which the molecules and their receptors become important regulators of the TME and major players in cancer biology. With the ability to activate certain signaling pathways, chemokine receptors may facilitate tumor progression at each of the key steps, including proliferation, angiogenesis, immunomodulation, and metastasis [13]. In addition, increasing studies have exhibited the property of chemokines in facilitating information exchanging between cancer cells and TME cells such as endothelial cells and fibroblasts, which in turn promotes the infiltration and activation of immune cells such as neutrophils and tumor-associated macrophages (TAMs) [28].
MVs and chemokines in TME
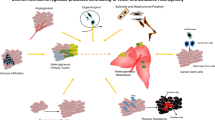
The molecular mechanisms underlying the functional interactions between cancer cells and the TME have been considered the subject of great moment. Historically these interactions are thought to be primarily mediated by signaling molecules such as cytokines and growth factors [33]. TME consists of various cell types, among them are fibroblasts, lymphocytes, inflammatory cells, epithelial cells, endothelial cells, and mesenchymal stem cells [34]. These cells interact with cancer cells and, together they form the intrinsic communication networks that affect several cancer hallmarks (Fig. 3). Studies indicate that such intercellular communications were modulated by various factors, such as growth factors, cytokines, and chemokines. Similar to these molecules, recent advances in cancer biology reveal that MVs also serve as a regulatory agent in such communications [11, 34].

Interaction between cancer cells and different components of the tumor microenvironment by the mediators of EVs
Peripheral blood system
In recent years, MVs have been described in cancer research as tumor-derived microvesicles (TMVs) [35]. Several reports documented that TMVs induce chemotaxis of leukocytes. Vesicles shed by cell lines of non-small-cell lung carcinoma, pancreatic adenocarcinoma, and colorectal adenocarcinoma stimulated chemotaxis of granulocytes, lymphocytes, and monocytes in vitro [36]. While in the plasma of hemophilia A patients, higher levels of MVs derived from endothelial cells, neutrophils, T lymphocytes, erythrocytes, and platelets were observed after exposure to exogenous FVIII, with distinct immunological profiles [37]. Human eosinophils could secrete cytokines, chemokines and cationic proteins, trafficking, and releasing them for roles in inflammation and other immune responses. When eosinophils are activated immediately after isolation and dissected by transmission electron microscopy, EVs are identified as MVs outwardly shedding off the plasma membrane. Both chemoattractant protein-11 (CCL11) and tumor necrosis factor-α (TNF-α) induce significant increase of MVs compared with non-activated cells [38].
Macrophage-monocyte system
TMVs carry several proteins and mRNA of tumor cells and can transfer some of them to monocytes. It is found that TMVs could activate monocytes, as evident by increased human leukocyte antigen-DR isotype (HLA-DR) expression, induce production of reactive oxygen intermediates (ROI), and mRNA accumulation and protein secretion of TNF, interleukin (IL)-10, IL-12p40 [36]. Furthermore, TMVs can exert anti-apoptotic effect on monocytes and activate serine/threonine kinase (AKT) by transferring CCR6 and CD44v7/8 to monocytes, altering immunologic phenotype and biological activity of the recipients [39]. TMVs induce expression of IL-8 (CXCL8), monocyte chemoattractant protein-1 (CCL2), macrophage inflammatory protein-1α (CCL3) and major intrinsic protein of lens fiber-1β (MIP-1β) (CCL4), and regulate on activation normal T-cells expressed and secreted CCL5 chemokines and accumulation of their mRNA in monocytes. Moreover, TMVs enhance angiogenesis in non-obese diabetic/severe combined immunodeficiency (NOD-SCID) mice by delivering chemokines and via stimulation of monocytes [40]. Monocytes are direct precursors of hematopoietic stem cell-derived macrophages. After their recruitment into the tumor tissue, they can differentiate into tumor-associated macrophages, and support tumor initiation, local progression, and distant metastasis [41]. It is becoming clear that macrophages, like other members of the myeloid family, are incredible heterogeneous and depending on tumor biology, different subpopulations of tumor associated macrophages may differ considerably in terms of function and phenotype [42]. In B16-F0 melanoma or EL-4 lymphoma cell lines, tumor cell-derived exosomes (TE) enhance the ability of mesenchymal stromal cells (MSCs) to promote macrophage infiltration. Ablation of macrophages by clodronate liposome administration reverses the tumor-promoting effect. In this process, TE-MSCs produce a large amount of CCR2 ligands, CCL2 and CCL7, which are responsible for macrophage recruitment [43].
Stromal cells
Increasing evidence has disclosed that stromal cells in the TME are also fundamental in tumor progression [44, 45]. Apart from extracellular matrix (ECM), TME includes non-malignant stromal cells surrounding the tumor cells, including fibroblasts, adipocytes, endothelial cells and inflammatory immune cells. The interplay between tumor cells and TME has been increasingly recognized as a principle determinant of malignancy. The stromal elements secrete chemokines functioning in a paracrine manner, which could induce ECM remodeling and enhance cancer proliferation and invasion. For example, we recently found that overexpression of the chemokines CXCL14 and CCL17 in mammary fibroblasts could enhance proliferation, migration, invasion of breast cancer epithelial cells, and contribute to chemo-resistance and disease relapse [45]. Chemokines of the CXCL family are present in the pancreatic TME and play a vital role in regulating PC progression. Most members of the chemokine family, including CXCL1, CXCL2, CXCL5, CXCL9, CXCL10 and CXCL13, where they are secreted by cancer or stromal cells, such as cancer-associated fibroblasts (CAFs) and dendritic cells (DCs). Most of these ligands have been reported to promote chemoresistance, immunosuppression, tumor proliferation and metastasis [46]. In the HIC1-deleted breast cancer cells, CXCL14 bound to its novel cognate receptor GPR85 on CAFs in the TME and was responsible for activating these fibroblasts via the extracellular regulated MAP kinase1/2 (ERK1/2), AKT, and neddylation pathways, promoting cancer progression via the induction of the epithelial-mesenchymal transition (EMT) by the CCL17/CCR4 axis [45].
Large oncosome-induced migration of CAFs can be potentiated by EVs derived from tumor cells in which miR-1227 has been overexpressed. Interestingly, this forced expression of the miRNA intracellularly results in a 3-fold change in large oncosomes in comparison to exosomes [47]. Large oncosomes can also potently stimulate expression of metastasis-associated factors, such as brain-derived neurotrophic factor (BDNF), CXCL12 and osteopontin, in stromal cells [24]. Normal T-cells secrete CCL5, which stimulates the externalization of S100 calcium binding protein A4 (S100A4) via MVs shedded from the plasma membrane of tumor and stromal cells. In wild type and S100A4-deficient mouse models, tumor cell-derived CCL5 on S100A4 release into blood circulation ultimately increases the metastatic burden in mice [48]. EVs produced by the highly metastatic rat pancreatic adenocarcinoma cell line BSp73AS preferentially target lung fibroblasts and lymph node stromal cells, triggering in these cells the upregulation or de novo expression of several adhesion molecules, chemokines, growth factors, and proteases, thus promoting pre-metastatic niche formation [49].
Dendritic-T cell system
TMVs are natural cargos for delivering tumor antigens and innate signals to DCs for tumor-specific T cell immunity. TMVs, once entering intestinal lumen, were mainly taken up by ileac intestinal epithelial cells (IECs), where TMVs activated nucleotide binding oligomerization domain containing 2 (NOD2) and its downstream mitogen activated kinase-like protein (MAPK) and nuclear factor-κB (NF-κB), leading to release of chemokines including CCL2, from IECs to attract CD103+ CD11c+ DCs, leading to subsequent antitumor T cell responses [50]. In the cohort of previously untreated hemophilia A patients, immunological profiles were distinct, higher levels of IL8, IL6, IL4, IL10, IL2, IL17A, and lower levels of CXCL10 and CCL2 were observed compared with non-haemophiliac cohorts. Also, higher levels of MVs derived from endothelial cells, neutrophils, T lymphocytes, erythrocytes, and platelets were observed [37]. Few leukemia-associated antigens (LAA) are characterized for acute myeloid leukemia (AML), apoptotic tumor cells constitute an attractive LAA source for personalized DC-based vaccines. DCs preferably ingest apoptotic blebs (MVs that require additional isolation steps) and are superior in migrating toward CCL19. Co-culturing bleb-loaded DCs with T cells led to an increased CD4+ T cell proliferation and increased interferon gamma (IFNγ) production by allogeneic T cells. Superior ingestion efficiency and migration, combined with favorable T cell cytokine release and CD8+ T cell priming ability and avidity, point to blebs as the preferred component of apoptotic leukemic cells for LAA loading of DC for the immunotherapy of AML [51]. Karin et al. demonstrated that CXCL10 acted on CD4+ and CD8+ T cells to enhance anti-tumor immunity, blocking the CCR8-CCL1 interaction, alone or combined with other immune checkpoint inhibitors, as an approach to treat malignant diseases [13]. Heat stressed tumor cells produce chemokine-enriched exosomes which are termed HS-TEX, which chemoattract and activate DCs and T cells more potently than conventional tumor-derived exosomes do. The enriched chemokines include CCL2, CCL3, CCL4, CCL5, and CCL20, enabling chemotaxis of DCs and T cells both in vitro and in vivo. Intratumoral injection of HS-TEX could induce specific antitumor immune response more efficiently than that by tumor-derived exosomes, inhibiting tumor growth and significantly prolonging survival of tumor-bearing mice. Therefore, heat stress may alter the functional attributes of tumor-derived exosomes, and the resulting HS-TEX may be an efficient tumor vaccine [52].
TMVs and chemokines in tumor progression
TMVs have emerged as essential mediators of cancer progression, which alter the metastatic behavior of primary tumors mainly through transport of their bioactive contents including oncogenes, oncoproteins, microRNAs, as well as transcripts of proteins and chemokines involved in angiogenesis or inflammation (Table 1). Moreover, TMVs have been shown to influence distant cellular niches, establishing favorable microenvionmental conditions that support growth of disseminated cancer cells upon their arrival at these pre-metastatic niches (Fig. 4).

Schematic of molecules and nucleic acids transfer by EVs in the process of cancer progression. Transmembrane and soluble proteins, lipids and nucleic acids are selectively coalesced into the multivesicular endosomes (MVEs) or into MVs shedding from the plasma membrane. MVEs fuse with the plasma membrane to secret exosomes into the extracellular environment. Exosomes and MVs may either be endocytosed by recipient cells or fuse directly with the plasma membrane. Both pathways result in the delivery of proteins, lipids and nucleic acids into the membrane or cytosol of the recipient cell to transfer the information, which potentially influence the progress of cancer development
Proliferation
Cancer cells release significantly larger amount of shedding MVs than their normal counterparts, which are associated with increased invasiveness and disease progression. Genetic alterations are needed to ensure sustained growth and proliferation of cancer cells and MVs facilitates intercellular spread of oncogenes, therefore enabling horizontal transfer of aggressive phenotypes. In gastric cancer patients, the amount of circulating MVs was elevated in all stages compared to normal people and, significantly higher in more advanced disease. MVs of these patients exhibited an increased expression of CCR6 and HER-2/neu on the membrane [53]. CD44H, CD44v6 and CCR6 molecules may play a role in attachment of TMVs to cancer cells, while HER-2 associated with CD24 may be involved in promoting growth of cancer cells. Pre-exposure of cancer cells to these TMVs resulted in enhancement of tumor growth and cancer cell-induced angiogenesis in vivo [5]. Interestingly, this signature presents important overlaps with other microenvironmental stimuli such as B-cell receptor stimulation, CLL/nurse-like cells co-culture or those provided by a lymph node microenvironment. EVs from MSCs of leukemic patients also rescue leukemic cells from spontaneous or drug-induced apoptosis, resulting in a higher migration and also a stronger gene modification [54].
Indolent glioma cells can acquire epidermal growth factor receptor variant III (EGFRvIII), a tumor-specific truncated form of EGFR, from microvesicles released by aggressive glioma cells harboring this variant [55]. Activation of growth promoting MAPK and AKT signaling pathways ensues, resulting in morphological transformation and anchorage-independent growth of the recipient tumor cells. In vitro studies confirmed that transfer of MV-encapsulated EGFRvIII mRNA also stimulated proliferation of glioblastoma cells [56], although it is unclear whether this signaling pathway is active in vivo. Interestingly, the uptake of TMVs is mediated by PS exposed on the surface, which can be blocked using Annexin V or EGFRvIII kinase inhibitors, further confirming the association between phenotypic switch and MVs transfer of oncoproteins or oncogenes. By in vitro and in vivo stem-like glioblastoma models, EVs isolated from glioblastoma-conditioned media with PKH67-label induce a proliferative phenotype in recipient glioblastoma cells. Using fluorescence activated cell-sorting analysis, the percentile of PKH67+ cells after incubation showed a sigmoidal log-linear dose-dependent relationship with the amount of PKH67-labelled EVs added. CCR8 acts as an EV receptor on glioblastoma cells and binds to CCL18, which acts as a bridging molecule. CCR8 inhibition caused a strong and consistent reduction in EVs uptake, neutralizes EVs-induced phenotypes in vitro [54].
In addition, MVs are capable of transitioning normal cells in TME into anaplastic cells. Antonyak et al. [57] demonstrated that MVs released by MDA-MB-231 human breast cancer cell line and U87 glioblastoma cell lines contain tissue transglutaminase (tTG), the protein cross-linking enzyme, and FN, the tTG-binding molecule and cross-linking substrate. Microvesicle-mediated transfer of cross-linked FN and tTG to recipient fibroblasts induces their transformation and aberrant proliferation by phosphorylation of focal adhesion kinase (FAK) and ERK kinases and activation of mitogenic signaling pathways.
Angiogenesis
Angiogenesis, the process involving formation of new blood vessels, is another hallmark of cancer and is of significance in promoting tumor dissemination and migration. Numerous studies have demonstrated that MVs can interact with endothelial cells, therefore stimulating angiogenic responses. TMVs harboring activated EGFR can be taken up by endothelial cells, leading to activation of MAPK and AKT signaling pathways, accompanied by increased expression of endogenous vascular endothelial growth factor (VEGF) and the autocrine activation of VEGF-2, the key receptor for VEGF signaling pathway [58]. In NSCLC, patient-derived circulating TMVs enhance vascular endothelial growth factor receptor 2 (VEGFR2) expression, as well as angiogenesis, nitric-oxide production, and endothelial cell proliferation. The amount of circulating MVs is highly correlated with pro-angiogenic factors at cellular and protein levels. In another study, rats treated with patient-derived circulating MVs exhibit higher microvessel count, more CXCR4+ and VEGF+ cells, and accelerate pulmonary metastatic hepatocellular carcinoma growth [59]. Endothelial cells that receive CD138 from multiple myeloma derived MVs are significantly stimulated so as to proliferate, secrete IL-6 and VEGF, two key angiogenic factors of myeloma, and form tubes in vitro and in vivo [60].
Aside from carrying bioactive EGFR variants, the aforementioned glioblastoma derived TMVs are loaded with angiogenic proteins, such as fibroblast growth factor (FGF), IL-6, and VEGF, which are capable of stimulating angiogenesis in vitro [57]. By advancing formation of new blood vessels in human brain microvascular endothelial cells, these TMVs were reported to stimulate cancer proliferation, motility, and tube formation in a dose-response manner [61]. Similarly, Hong et al. [62] identified 241 mRNAs, which were enriched in colorectal cancer cell-derived MVs. Treatment of endothelial cells with these MVs resulted in a significant increase in proliferation, which is in line with the results of network analysis. MVs shed from CD105+ human renal cancer stem cells confer an activated angiogenic phenotype to normal epithelial cells, stimulate blood vessel formation after in vivo implantation in severe combined immunodeficient (SCID) mice, and enhance risk of develo** lung metastases [63]. Besides pro-angiogenic growth factors, proteinases and cytokines, microvesicles may cargo miRNAs to mediate angiogenesis, as is exemplified in gastric cancer [64] and colorectal cancer [65].
Metastasis
The invasive and migratory properties of tumor cells accumulate when tumor cells grow and evolve [11]. This hallmark of cancer is associated with MV-encapsulating proteases, such as the MMP family. This family of enzymes can degrade ECM and catalyze the proteolysis of the basement membrane, therefore enhancing the mobility of migrating tumor cells and allowing them to enter the circulatory system. In amoeboid-like invasive melanoma cell lines, vesicular soluble N-ethylmaleimide-sensitive factor attachment protein receptor (v-SNARE) and vesicle-associated membrane protein 3 (VAMP3) have been identified as two key regulators for delivery of the cargo molecules to shedding TMVs, such as the membrane-type 1 matrix metalloprotease (MT1-MMP). These TMVs markedly facilitate the maintenance of amoeboid phenotype and allow for cell invasion. VAMP3-shRNA transfected cells are lacking in TMVs that contain MT1-MMP, making them difficult to invade through dense and highly cross-linked matrices such as rat-tail collagen compared to those transfected with scramble shRNA [4]. VMR, CSML100, and CSML0 mouse adenocarcinoma cell lines originated from two independent spontaneous tumors in A/Sn mice, CCL5 stimulates the externalization of S100A4 via TMVs shedding from the plasma membrane of these tumor and stroma cells, which in its turn induces the upregulation of FN in fibroblasts and a number of cytokines in tumor cells including CCL5 [48]. In TMVs ARF, modulation of ARF1 expression dramatically impairs the ability of MDA-MB-231 cells to degrade the extracellular matrix by adjusting MMP9 activity, to inhibit invasiveness and metastasis [66, 67].
In prostate cancer (PCa) cell lines with mesenchymal characteristics (22Rv1/CR-1; Mes-PCa), TMVs were found to promote and maintain mesenchymal features in the recipient epithelium-like prostate cancer cells, modulating androgen receptor signaling and activating transforming growth factor beta (TGF-β) signaling pathway in the meantime. Moreover, these recipient cells which have attained mesenchymal traits exhibited enhanced migratory and invasive potentials, as well as increased resistance to the androgen receptor antagonist enzalutamide [68]. CXCR7 is a chemokine that has been proven responsible for PCa progression. As a direct downstream target of hypermethylated in cancer 1 gene (HIC1), restoring HIC1 expression in PCa cells markedly inhibited proliferation, migration, and invasion and induced the apoptosis in these cells [51]. Studies by the Rughetti group [90, 91] revealed that microvesicle-mediated antigen transfer to DCs is of crucial importance for cross-presentation of tumor-glycosylated antigens. In particular, mucine 1 (MUC1), one of the most relevant glycoproteins associated with carcinogenesis, was cross-processed and presented to antigen-specific CD8+ T cells when carried by MVs, while the internalized soluble form of MUC1 was retained in the endolysomal/HLA-II compartment and did not activate any T cell response. They further proposed that the controversial roles of MVs in modulating immunity are dependent upon the stage of tumor progression.
DC-derived exosomes contain series of costimulatory molecules including B7–1 (CD80), B7–2 (CD86), programmed death 1-ligand (PD-L1) and PD-L2. Rather than PD-L1 and PD-L2, therapeutic effects of IL-10 treated DC and exosomes required both B7–1 and B7–2, which play a critical role in immunosuppressive functions of both DC and exosomes, giving the growing interest in exosomes for therapeutic applications [92]. In glioblastoma, PD-L1 was expressed on the surface of some glioblastoma-derived EVs, with the potential to directly bind to programmed death-1 (PD1). These EVs block T cell activation and proliferation in response to T cell receptor stimulation. Blocking PD1 pathway significantly reversed the EV-mediated blockade of T cell activation but only when PD-L1 was present on EVs. When glioblastoma PD-L1 was up-regulated by IFN-γ, EVs also showed some PD-L1 dependent inhibition of T cell activation [93]. HER2-positive breast cancer cells with stable over-expressing Neuromedin U and their released EVs have increased amounts of the immunosuppressive cytokine TGFβ1 and the lymphocyte activation inhibitor PD-L1, show enhanced resistance to antibody-dependent cell cytotoxicity mediated by trastuzumab, indicating a role of Neuromedin U in enhancing immune evasion [94]. While in malignant glioma, monocytes from naïve patient peripheral blood treatmented with glioma-derived exosomes fail to induce monocytic PD-L1 expression or alter the activation of cytotoxic T-cells, but promote immunosuppressive HLA-DR low monocytic phenotypes [95].
Probably the most promising future for therapeutic use of MVs in cancer immunotherapy is to be administered as vaccines. In their study, Zhang et al. [96] immunized mice with extracellular vesicles isolated from different cancer cell lines, and as a result, 50% of the microparticle-immunized mice remained tumor-free after injected tumor challenges. They further discovered that tumor-derived microvesicles confer DNA fragments to DCs, leading to type I IFN production through the cGAS/STING-mediated DNA-sensing pathway. Type I IFN, in its turn, stimulate DC’s maturation and antigen-presenting capabilities. Notably, Zhang et al. reported a much lower 12.5% tumor-free rate of exosome-immunized mice after the tumor challenges. This suggests that, although the present development of extracellular vesicle-based vaccines is largely focusing on exosomal vaccines, microparticle-based vaccines appear to be more immunogenic.
Taken together, these studies highlight the potential clinical applicability of microvesicle-based vaccines in cancer immunotherapy. In future, these vaccines are expected to be administered alongside immune checkpoint inhibitors, the currently well-established immunotherapeutic approach, to further augment anti-tumor immunity.
Conclusions
As evidence has been shown from the literature, MVs are extensively studied and greatly contribute to the pathogenesis of multiple cancer types. With the growing understanding of the biology and biogenesis of MVs in cancer pathophysiology, MV research has been spawning much excitement in the past decade. Of particular interest for the current discussion is the intercellular communication between cancer cells and stromal cells in TME, which frequently involves bidirectional transfer of encapsulated chemokines. Presently available studies have looked at TMVs using in vitro cell lines or in vivo animal models, and revealed the importance of MVs as key mediators of cancer growth, proliferation, apoptosis, angiogenesis, coagulation and metastasis, proposing a paradigm shift of using TMVs as diagnostic or prognostic biomarkers. Moreover, TMVs have been shown to contribute to chemo-resistance and immunomodulation of cancer cells, shedding light on the clinical application of TMV-based or TMV-targeted therapeutic interventions to augment the efficacy of chemotherapy or immunotherapy. Nevertheless, current understanding of the TMVs and TMV-related chemokines has only scratched the surface. In order to demonstrate the authentic physiological functions of MVs in vivo, it is imperative to design animal models in which release and uptake of chemokine-containing MVs can be specifically monitored and interfered. With the mechanism of how MVs mediate intercellular communication becomes increasingly appreciated, MVs are expected to reshape our view towards cancer biology, become important component in laboratory research and elucidate novel therapeutic strategies for various cancer types.
Abbreviations
- AKT:
-
Serine/threonine kinase
- AML:
-
Acute myeloid leukemia
- ARF6:
-
ADP-ribosylation factor 6
- BCRP:
-
Breast cancer resistance protein
- BDNF:
-
Brain-derived neurotrophic factor
BM
Bone marrow
- CAF:
-
Cancer-associated fibroblast
- CD:
-
Cluster of differentiation
- CHAMP3:
-
Charged multivesicular body protein 3
- CLL:
-
Chronic lymphocytic leukemia
- DC:
-
Dendritic cell
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- ERK:
-
Extracellular regulated MAP kinase
- ES:
-
Embryonic stem
- EV:
-
Extracellular vesicles
- FAK:
-
Focal adhesion kinase
- FN:
-
Fibronectin
- HIC-1:
-
Hypermethylated in cancer 1
- HLA-DR:
-
Human leukocyte antigen-DR isotype
- HS-TEX:
-
Heat-stressed tumor cells
- IAP:
-
Inhibitors of apoptosis protein
- IEC:
-
Intestinal epithelial cell
- IFNγ:
-
Interferon gamma
- IL:
-
Interleukin
- LAA:
-
Leukemia-associated antigen
- MAPK:
-
Mitogen activated kinase-like protein
- MIP-1β:
-
Major intrinsic protein of lens fiber-1β
- MMP:
-
Matrix metalloproteinase
- MRP1:
-
Multidrug resistance-associated protein 1
- NF-κB:
-
Nuclear factor-κB
- NOD2:
-
Nucleotide binding oligomerization domain containing 2
- MSC:
-
Mesenchymal stromal cell
- MT1-MMP:
-
Membrane-type 1 matrix metalloprotease
- MUC1:
-
Mucine 1
- MVE:
-
Multivesicular endosome
- MV:
-
Microvesicle
- PD1:
-
Programmed death 1
- PD-L1:
-
Programmed death 1-ligand
- P-gp:
-
P-glycoprotein
- PS:
-
Phosphatidylserine
- ROI:
-
reactive oxygen intermediates
- S100A4:
-
S100 calcium binding protein A4
- SCID:
-
Severe combined immunodeficient
- TAM:
-
Tumor-associated macrophages
- TE:
-
Tumor cell-derived exosomes
- TGFβ:
-
Transforming growth factor beta
- TNF-α:
-
Tumor necrosis factor-α
- TME:
-
Tumor microenvironment
- TMV:
-
Tumor-derived microvesicles
- TMZ:
-
Temozolomide
- tTG:
-
Tissue transglutaminase
- PCa:
-
Prostate cancer
- uPA:
-
Urokinase plasminogen activator
- VAMP-3:
-
Vesicle-associated membrane protein 3
- VEGF:
-
Vascular epithelium growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
- v-SNARE:
-
Vesicular soluble N-ethylmaleimide-sensitive factor attachment protein receptor
References
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83.
Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20(5):847–56.
Desrochers LM, Bordeleau F, Reinhart-King CA, Cerione RA, Antonyak MA. Microvesicles provide a mechanism for intercellular communication by embryonic stem cells during embryo implantation. Nat Commun. 2016;7:11958.
Clancy JW, Sedgwick A, Rosse C, Muralidharan-Chari V, Raposo G, Method M, et al. Regulated delivery of molecular cargo to invasive tumour-derived microvesicles. Nat Commun. 2015;6:6919.
Stec M, Szatanek R, Baj-Krzyworzeka M, Baran J, Zembala M, Barbasz J, et al. Interactions of tumour-derived micro (nano) vesicles with human gastric cancer cells. J Transl Med. 2015;13:376.
D'souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26(12):1287–99.
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–15.
Holm MM, Kaiser J, Schwab ME. Extracellular vesicles: multimodal envoys in neural maintenance and repair. Trends Neurosci. 2018;41(6):360–72.
Willms E, Cabanas C, Mager I, Wood MJA, Vader P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in Cancer progression. Front Immunol. 2018;9:738.
Barteneva NS, Fasler-Kan E, Bernimoulin M, Stern JN, Ponomarev ED, Duckett L, et al. Circulating microparticles: square the circle. BMC Cell Biol. 2013;14:23.
Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41–51.
Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, De Candia P, et al. Focus on extracellular vesicles: new Frontiers of cell-to-cell communication in Cancer. Int J Mol Sci. 2016;17(2):175.
Karin N. Chemokines and cancer: new immune checkpoints for cancer therapy. Curr Opin Immunol. 2018;51:140–5.
Weitzenfeld P, Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett. 2014;352(1):36–53.
Wendler F, Favicchio R, Simon T, Alifrangis C, Stebbing J, Giamas G. Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene. 2017;36(7):877–84.
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20(9):1487–95.
Surman M, Stepien E, Hoja-Lukowicz D, Przybylo M. Deciphering the role of ectosomes in cancer development and progression: focus on the proteome. Clin Exp Metastasis. 2017;34(3–4):273–89.
Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31(1):15–26.
Pap E, Pallinger E, Pasztoi M, Falus A. Highlights of a new type of intercellular communication: microvesicle-based information transfer. Inflamm Res. 2009;58(1):1–8.
Agrahari V, Agrahari V, Burnouf PA, Chew CH, Burnouf T. Extracellular microvesicles as new industrial therapeutic Frontiers. Trends Biotechnol. 2019. https://doi.org/10.1016/j.tibtech.2018.11.012.
Turiak L, Misjak P, Szabo TG, Aradi B, Paloczi K, Ozohanics O, et al. Proteomic characterization of thymocyte-derived microvesicles and apoptotic bodies in BALB/c mice. J Proteome. 2011;74(10):2025–33.
Lane JD, Allan VJ, Woodman PG. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J Cell Sci. 2005;118(Pt 17):4059–71.
Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69(13):5601–9.
Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181(5):1573–84.
Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–7.
Namas RA, Mi Q, Namas R, Almahmoud K, Zaaqoq AM, Abdul-Malak O, et al. Insights into the role of chemokines, damage-associated molecular patterns, and lymphocyte-derived mediators from computational models of trauma-induced inflammation. Antioxid Redox Signal. 2015;23(17):1370–87.
Rudemiller NP, Crowley SD. The role of chemokines in hypertension and consequent target organ damage. Pharmacol Res. 2017;119:404–11.
Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29(4):709–22.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72.
Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018. https://doi.org/10.1111/febs.14466.
Chen K, Bao Z, Tang P, Gong W, Yoshimura T, Wang JM. Chemokines in homeostasis and diseases. Cell Mol Immunol. 2018;15(4):324–34.
Idorn M, Thor SP. Chemokine receptors and exercise to tackle the inadequacy of T cell homing to the tumor site. Cells. 2018. https://doi.org/10.3390/cells7080108.
Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene. 2015;34(3):290–302.
Emon B, Bauer J, Jain Y, Jung B, Saif T. Biophysics of tumor microenvironment and Cancer metastasis - a mini review. Comput Struct Biotechnol J. 2018;16:279–87.
Whiteside TL. Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Br J Cancer. 2005;92(2):209–11.
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Zembala M. Tumour-derived microvesicles modulate biological activity of human monocytes. Immunol Lett. 2007;113(2):76–82.
Jardim LL, Chaves DG, Silveira-Cassette ACO, Simoes ESAC, Santana MP, Cerqueira MH, et al. Immune status of patients with haemophilia a before exposure to factor VIII: first results from the HEMFIL study. Br J Haematol. 2017;178(6):971–8.
Akuthota P, Carmo LA, Bonjour K, Murphy RO, Silva TP, Gamalier JP, et al. Extracellular microvesicle production by human eosinophils activated by "inflammatory" stimuli. Front Cell Dev Biol. 2016;4:117.
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Urbanowicz B, Branski P, et al. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol Immunother. 2006;55(7):808–18.
Baj-Krzyworzeka M, Weglarczyk K, Mytar B, Szatanek R, Baran J, Zembala M. Tumour-derived microvesicles contain interleukin-8 and modulate production of chemokines by human monocytes. Anticancer Res. 2011;31(4):1329–35.
Richards DM, Hettinger J, Feuerer M. Monocytes and macrophages in cancer: development and functions. Cancer Microenviron. 2013;6(2):179–91.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Lin LY, Du LM, Cao K, Huang Y, Yu PF, Zhang LY, et al. Tumour cell-derived exosomes endow mesenchymal stromal cells with tumour-promotion capabilities. Oncogene. 2016;35(46):6038–42.
Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32.
Wang Y, Weng X, Wang L, Hao M, Li Y, Hou L, et al. HIC1 deletion promotes breast cancer progression by activating tumor cell/fibroblast crosstalk. J Clin Invest. 2018;128(12):5235–50.
Lee NH, Nikfarjam M, He H. Functions of the CXC ligand family in the pancreatic tumor microenvironment. Pancreatology. 2018;18(7):705–16.
Morello M, Minciacchi VR, De Candia P, Yang J, Posadas E, Kim H, et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle. 2013;12(22):3526–36.
Forst B, Hansen MT, Klingelhofer J, Moller HD, Nielsen GH, Grum-Schwensen B, et al. Metastasis-inducing S100A4 and RANTES cooperate in promoting tumor progression in mice. PLoS One. 2010;5(4):e10374.
Rana S, Yue S, Stadel D, Zoller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012;44(9):1574–84.
Dong W, Zhang H, Yin X, Liu Y, Chen D, Liang X, et al. Oral delivery of tumor microparticle vaccines activates NOD2 signaling pathway in ileac epithelium rendering potent antitumor T cell immunity. Oncoimmunology. 2017;6(3):e1282589.
Ruben JM, Van Den Ancker W, Bontkes HJ, Westers TM, Hooijberg E, Ossenkoppele GJ, et al. Apoptotic blebs from leukemic cells as a preferred source of tumor-associated antigen for dendritic cell-based vaccines. Cancer Immunol Immunother. 2014;63(4):335–45.
Chen T, Guo J, Yang M, Zhu X, Cao X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J Immunol. 2011;186(4):2219–28.
Baran J, Baj-Krzyworzeka M, Weglarczyk K, Szatanek R, Zembala M, Barbasz J, et al. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol Immunother. 2010;59(6):841–50.
Berenguer J, Lagerweij T, Zhao XW, Dusoswa S, Van Der Stoop P, Westerman B, et al. Glycosylated extracellular vesicles released by glioblastoma cells are decorated by CCL18 allowing for cellular uptake via chemokine receptor CCR8. J Extracell Vesicles. 2018;7(1):1446660.
Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10(5):619–24.
Skog J, Wurdinger T, Van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6.
Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, et al. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A. 2011;108(12):4852–7.
Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A. 2009;106(10):3794–9.
Ko SF, Yip HK, Zhen YY, Lee CC, Li JH, Lee CC, et al. Cancer patient-derived circulating microparticles enhance lung metastasis in a rat model: dual-source CT, cellular, and molecular studies. Mol Imaging Biol. 2016;18(4):490–9.
Liu Y, Zhu XJ, Zeng C, Wu PH, Wang HX, Chen ZC, et al. Microvesicles secreted from human multiple myeloma cells promote angiogenesis. Acta Pharmacol Sin. 2014;35(2):230–8.
Giusti I, Delle Monache S, Di Francesco M, Sanita P, D'ascenzo S, Gravina GL, et al. From glioblastoma to endothelial cells through extracellular vesicles: messages for angiogenesis. Tumour Biol. 2016;37(9):12743–53.
Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics. 2009;10:556.
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71(15):5346–56.
Zhang H, Bai M, Deng T, Liu R, Wang X, Qu Y, et al. Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett. 2016;375(2):331–9.
Yamada N, Tsujimura N, Kumazaki M, Shinohara H, Taniguchi K, Nakagawa Y, et al. Colorectal cancer cell-derived microvesicles containing microRNA-1246 promote angiogenesis by activating Smad 1/5/8 signaling elicited by PML down-regulation in endothelial cells. Biochim Biophys Acta. 2014;1839(11):1256–72.
Schlienger S, Campbell S, Claing A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol Biol Cell. 2014;25(1):17–29.
Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31(45):4740–9.
El-Sayed IY, Daher A, Destouches D, Firlej V, Kostallari E, Maille P, et al. Extracellular vesicles released by mesenchymal-like prostate carcinoma cells modulate EMT state of recipient epithelial-like carcinoma cells through regulation of AR signaling. Cancer Lett. 2017;410:100–11.
Zheng J, Wang J, Sun X, Hao M, Ding T, **ong D, et al. HIC1 modulates prostate cancer progression by epigenetic modification. Clin Cancer Res. 2013;19(6):1400–10.
Wang J, Shiozawa Y, Wang J, Wang Y, Jung Y, Pienta KJ, et al. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J Biol Chem. 2008;283(7):4283–94.
Kalinkovich A, Tavor S, Avigdor A, Kahn J, Brill A, Petit I, et al. Functional CXCR4-expressing microparticles and SDF-1 correlate with circulating acute myelogenous leukemia cells. Cancer Res. 2006;66(22):11013–20.
Jaszczynska-Nowinka K, Rucinski M, Ziolkowska A, Markowska A, Malendowicz LK. Expression of SDF-1 and CXCR4 transcript variants and CXCR7 in epithelial ovarian cancer. Oncol Lett. 2014;7(5):1618–24.
Lima LG, Leal AC, Vargas G, Porto-Carreiro I, Monteiro RQ. Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb Res. 2013;132(4):450–6.
Pang A, Cui Y, Chen Y, Cheng N, Delaney MK, Gu M, et al. Shear-induced integrin signaling in platelet phosphatidylserine exposure, microvesicle release, and coagulation. Blood. 2018;132(5):533–43.
Crompot E, Van Damme M, Pieters K, Vermeersch M, Perez-Morga D, Mineur P, et al. Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica. 2017;102(9):1594–604.
Liebhardt S, Ditsch N, Nieuwland R, Rank A, Jeschke U, Von Koch F, et al. CEA-, Her2/neu-, BCRP- and Hsp27-positive microparticles in breast cancer patients. Anticancer Res. 2010;30(5):1707–12.
Lu JF, Luk F, Gong J, Jaiswal R, Grau GE, Bebawy M. Microparticles mediate MRP1 intercellular transfer and the re-templating of intrinsic resistance pathways. Pharmacol Res. 2013;76:77–83.
Wei Y, Lai X, Yu S, Chen S, Ma Y, Zhang Y, et al. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res Treat. 2014;147(2):423–31.
Jaiswal R, Luk F, Dalla PV, Grau GE, Bebawy M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS One. 2013;8(4):e61515.
De Souza PS, Cruz AL, Viola JP, Maia RC. Microparticles induce multifactorial resistance through oncogenic pathways independently of cancer cell type. Cancer Sci. 2015;106(1):60–8.
Shedden K, **e XT, Chandaroy P, Chang YT, Rosania GR. Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res. 2003;63(15):4331–7.
Jorfi S, Ansa-Addo EA, Kholia S, Stratton D, Valley S, Lange S, et al. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci Rep. 2015;5:13006.
Kholia S, Jorfi S, Thompson PR, Causey CP, Nicholas AP, Inal JM, et al. A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J Extracell Vesicles. 2015;4:26192.
Ma J, Zhang Y, Tang K, Zhang H, Yin X, Li Y, et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016;26(6):713–27.
Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68(4):1228–35.
Napoletano C, Rughetti A, Landi R, Pinto D, Bellati F, Rahimi H, et al. Immunogenicity of Allo-vesicle carrying ERBB2 tumor antigen for dendritic cell-based anti-tumor immunotherapy. Int J Immunopathol Pharmacol. 2009;22(3):647–58.
Moore C, Kosgodage U, Lange S, Inal JM. The emerging role of exosome and microvesicle- (EMV-) based cancer therapeutics and immunotherapy. Int J Cancer. 2017;141(3):428–36.
Maus RLG, Jakub JW, Nevala WK, Christensen TA, Noble-Orcutt K, Sachs Z, et al. Human melanoma-derived extracellular vesicles regulate dendritic cell maturation. Front Immunol. 2017;8:358.
Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66(18):9290–8.
Rughetti A, Rahimi H, Belleudi F, Napoletano C, Battisti F, Zizzari IG, et al. Microvesicle cargo of tumor-associated MUC1 to dendritic cells allows cross-presentation and specific carbohydrate processing. Cancer Immunol Res. 2014;2(2):177–86.
Battisti F, Napoletano C, Rahimi Koshkaki H, Belleudi F, Zizzari IG, Ruscito I, et al. Tumor-derived microvesicles modulate antigen cross-processing via reactive oxygen species-mediated Alkalinization of Phagosomal compartment in dendritic cells. Front Immunol. 2017;8:1179.
Ruffner MA, Kim SH, Bianco NR, Francisco LM, Sharpe AH, Robbins PD. B7-1/2, but not PD-L1/2 molecules, are required on IL-10-treated tolerogenic DC and DC-derived exosomes for in vivo function. Eur J Immunol. 2009;39(11):3084–90.
Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv. 2018;4;(3):eaar2766.
Martinez VG, O'neill S, Salimu J, Breslin S, Clayton A, Crown J, et al. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology. 2017;6(12):e1362530.
Iorgulescu JB, Ivan ME, Safaee M, Parsa AT. The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J Neuroimmunol. 2016;290:103–8.
Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res. 2015;3(2):196–205.
Acknowledgements
We thank Brandi Miller for the helps of this review.
Funding
Research in the authors’ laboratory is supported by the National Natural Funding of China (81402093, 81272404), National key program (973) for Basic Research of China (2011CB510106), the Program for Professor of Special Appointment (Eastern Scholar to J, Wang) at Shanghai Institutions of Higher Learning.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
XJB, YTX, TQW, MFY and LLD wrote the review; XJB prepared the figures; JHW and SCR conceived, provided direction, made critical revisions and proofread the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consented for publication in its present form.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bian, X., **ao, YT., Wu, T. et al. Microvesicles and chemokines in tumor microenvironment: mediators of intercellular communications in tumor progression. Mol Cancer 18, 50 (2019). https://doi.org/10.1186/s12943-019-0973-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-0973-7